
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.3131C>A | P1044Q 2D  AIThe SynGAP1 missense variant P1044Q is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -4.439 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.338 | Likely Benign | 0.51 | Neutral | 0.004 | Benign | 0.015 | Benign | 5.43 | Benign | 0.45 | Tolerated | 0.1544 | 0.5591 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3340A>T | S1114C 2D  AIThe SynGAP1 missense variant S1114C is reported in gnomAD (ID 6‑33443892‑A‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | 6-33443892-A-T | -8.600 | Likely Pathogenic | 0.091 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -1.77 | Neutral | 0.938 | Possibly Damaging | 0.552 | Possibly Damaging | 2.63 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1101 | 0.5675 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3368G>T | G1123V 2D  AIThe SynGAP1 missense variant G1123V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that G1123V is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | -7.129 | In-Between | 0.091 | Likely Benign | Likely Benign | 0.333 | Likely Benign | -1.03 | Neutral | 0.292 | Benign | 0.157 | Benign | 4.35 | Benign | 0.29 | Tolerated | 0.1362 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3854C>T | P1285L 2D  AIThe SynGAP1 missense variant P1285L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification for P1285L, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.821643 | Binding | 0.557 | 0.759 | 0.750 | -3.663 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.072 | Likely Benign | 0.06 | Neutral | 0.072 | Benign | 0.029 | Benign | 4.31 | Benign | 1.00 | Tolerated | 0.2159 | 0.4539 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3952C>G | L1318V 2D  AIThe SynGAP1 missense variant L1318V is catalogued in gnomAD (ID 6‑33451826‑C‑G) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence overwhelmingly supports a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this assessment does not contradict the ClinVar record, which contains no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.968271 | Binding | 0.399 | 0.865 | 0.750 | 6-33451826-C-G | 1 | 6.31e-7 | -3.938 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -0.58 | Neutral | 0.113 | Benign | 0.028 | Benign | 4.08 | Benign | 0.75 | Tolerated | 3.77 | 5 | 0.1861 | 0.3027 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.4018A>T | T1340S 2D  AIThe SynGAP1 missense variant T1340S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.977899 | Binding | 0.444 | 0.697 | 0.750 | -2.698 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.116 | Likely Benign | 0.62 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.87 | Benign | 1.00 | Tolerated | 0.3436 | 0.4521 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.4019C>G | T1340S 2D AIThe SynGAP1 missense variant T1340S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.977899 | Binding | 0.444 | 0.697 | 0.750 | -2.698 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.083 | Likely Benign | 0.62 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.87 | Benign | 1.00 | Tolerated | 0.3436 | 0.4521 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.112C>G | P38A 2D  AIThe SynGAP1 missense variant P38A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P38A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -3.179 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -2.03 | Neutral | 0.805 | Possibly Damaging | 0.857 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.3888 | 0.5977 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.1135T>A | S379T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S379T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are Rosetta, polyPhen‑2 HumDiv, and the Foldetta stability method. FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as pathogenic. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.433206 | Uncertain | 0.327 | 0.931 | 0.625 | -5.646 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 1.42 | Ambiguous | 0.6 | 3.96 | Destabilizing | 2.69 | Destabilizing | 0.06 | Likely Benign | 0.230 | Likely Benign | -0.50 | Neutral | 0.462 | Possibly Damaging | 0.084 | Benign | 3.87 | Benign | 0.16 | Tolerated | 0.2293 | 0.6248 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1909T>A | S637T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -4.116 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.1 | -0.91 | Ambiguous | -0.28 | Likely Benign | -0.39 | Likely Benign | 0.067 | Likely Benign | -0.19 | Neutral | 0.086 | Benign | 0.019 | Benign | 3.45 | Benign | 0.19 | Tolerated | 0.1675 | 0.4572 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1921T>G | S641A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S641A is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined (REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly classifies the substitution as benign. No pathogenic predictions are present. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.125101 | Structured | 0.157322 | Uncertain | 0.786 | 0.270 | 0.000 | -6.103 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.2 | -0.28 | Likely Benign | 0.05 | Likely Benign | 0.27 | Likely Benign | 0.067 | Likely Benign | -2.07 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.43 | Benign | 0.17 | Tolerated | 0.4873 | 0.4680 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.2027G>C | S676T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and Rosetta. No tool predicts a pathogenic outcome; the only inconclusive results are from FoldX (Uncertain) and Foldetta (Uncertain). High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta remains Uncertain. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -5.621 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.79 | Ambiguous | 0.1 | 0.34 | Likely Benign | 0.57 | Ambiguous | -0.36 | Likely Benign | 0.098 | Likely Benign | 0.91 | Neutral | 0.050 | Benign | 0.017 | Benign | 3.52 | Benign | 1.00 | Tolerated | 0.1575 | 0.6286 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2254T>G | S752A 2D AIThe SynGAP1 missense variant S752A has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -3.258 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -1.42 | Neutral | 0.910 | Possibly Damaging | 0.524 | Possibly Damaging | 1.59 | Pathogenic | 0.04 | Affected | 0.5092 | 0.5634 | Strenghten | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.22A>T | I8F 2D  AIThe SynGAP1 missense variant I8F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -3.000 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.29 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.02 | Benign | 0.00 | Affected | 0.0483 | 0.2871 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2564T>C | L855P 2D  AIThe SynGAP1 missense variant L855P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.485558 | Uncertain | 0.285 | 0.823 | 0.625 | -2.434 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -1.19 | Neutral | 0.586 | Possibly Damaging | 0.377 | Benign | 3.97 | Benign | 0.14 | Tolerated | 0.3633 | 0.1805 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2581T>G | S861A 2D AIThe SynGAP1 missense variant S861A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.540903 | Binding | 0.285 | 0.797 | 0.250 | -5.059 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -1.02 | Neutral | 0.409 | Benign | 0.172 | Benign | 4.08 | Benign | 0.48 | Tolerated | 0.4541 | 0.5401 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2828G>T | G943V 2D AIThe SynGAP1 missense variant G943V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The only tool with an uncertain outcome is ESM1b, which does not alter the overall consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign classification. High‑accuracy predictors corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Taken together, the evidence overwhelmingly supports a benign interpretation, and there is no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | -7.658 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.265 | Likely Benign | -0.43 | Neutral | 0.224 | Benign | 0.062 | Benign | 2.85 | Benign | 0.16 | Tolerated | 0.1399 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2848G>T | G950C 2D  AIThe SynGAP1 missense variant G950C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (five pathogenic vs. four benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -9.589 | Likely Pathogenic | 0.092 | Likely Benign | Likely Benign | 0.395 | Likely Benign | -0.35 | Neutral | 0.983 | Probably Damaging | 0.750 | Possibly Damaging | 2.26 | Pathogenic | 0.01 | Affected | 0.1406 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2861C>T | P954L 2D AIThe SynGAP1 missense variant P954L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984159 | Disordered | 0.932268 | Binding | 0.465 | 0.926 | 0.750 | -5.607 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.43 | Neutral | 0.977 | Probably Damaging | 0.812 | Possibly Damaging | 2.78 | Benign | 0.55 | Tolerated | 0.2346 | 0.5867 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2891A>T | H964L 2D  AIThe SynGAP1 missense variant H964L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.568 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.20 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.15 | Benign | 0.02 | Affected | 0.1409 | 0.5195 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.308G>C | G103A 2D  AIThe SynGAP1 missense variant G103A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.549 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.08 | Neutral | 0.008 | Benign | 0.008 | Benign | 4.31 | Benign | 0.00 | Affected | 0.3941 | 0.3784 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3197C>T | P1066L 2D AIThe SynGAP1 missense variant P1066L is listed in ClinVar as a benign variant (ClinVar ID 951518.0) and is present in gnomAD (ID 6‑33443749‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, which is consistent with the ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | Likely Benign | 1 | 6-33443749-C-T | 14 | 8.71e-6 | -5.478 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -3.68 | Deleterious | 0.996 | Probably Damaging | 0.903 | Possibly Damaging | 2.72 | Benign | 0.00 | Affected | 4.32 | 2 | 0.2269 | 0.6780 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.370G>C | A124P 2D  AIThe SynGAP1 missense variant A124P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A124P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.699139 | Binding | 0.340 | 0.883 | 0.750 | -3.030 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -1.19 | Neutral | 0.984 | Probably Damaging | 0.690 | Possibly Damaging | 4.05 | Benign | 0.03 | Affected | 0.2293 | 0.6141 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3728A>T | Q1243L 2D  AIThe SynGAP1 missense variant Q1243L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.545602 | Disordered | 0.433693 | Uncertain | 0.887 | 0.551 | 0.500 | -6.092 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -4.00 | Deleterious | 0.912 | Possibly Damaging | 0.629 | Possibly Damaging | 2.65 | Benign | 0.02 | Affected | 0.0541 | 0.3364 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3838A>C | M1280L 2D  AIThe SynGAP1 missense variant M1280L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for M1280L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -1.391 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.204 | Likely Benign | -2.23 | Neutral | 0.052 | Benign | 0.017 | Benign | 2.40 | Pathogenic | 0.00 | Affected | 0.1113 | 0.3186 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3838A>T | M1280L 2D AIThe SynGAP1 missense variant M1280L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar claim exists for this variant. Thus, based on current computational predictions, the M1280L variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -1.391 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.204 | Likely Benign | -2.23 | Neutral | 0.052 | Benign | 0.017 | Benign | 2.40 | Pathogenic | 0.00 | Affected | 0.1113 | 0.3186 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.4022C>G | A1341G 2D  AIThe SynGAP1 missense variant A1341G is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools overwhelmingly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only the SIFT algorithm predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Taken together, the preponderance of evidence points to a benign effect for A1341G, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this assessment does not contradict the ClinVar record, which contains no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.980111 | Binding | 0.383 | 0.696 | 1.000 | -3.334 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -0.84 | Neutral | 0.006 | Benign | 0.011 | Benign | 4.12 | Benign | 0.05 | Affected | 0.2026 | 0.4541 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1325A>G | K442R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K442R is catalogued in gnomAD (ID 6‑33438230‑A‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the only tools predicting a pathogenic outcome are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) classifies the variant as benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | 6-33438230-A-G | 1 | 6.20e-7 | -5.761 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.1 | 0.29 | Likely Benign | 0.21 | Likely Benign | 0.51 | Ambiguous | 0.098 | Likely Benign | -1.42 | Neutral | 0.972 | Probably Damaging | 0.875 | Possibly Damaging | 3.46 | Benign | 0.35 | Tolerated | 3.37 | 29 | 0.3885 | 0.0778 | 2 | 3 | -0.6 | 28.01 | ||||||||||||||||||||||||
| c.1412C>G | S471C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471C is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33438444‑C‑G). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of predictions (10 benign vs. 3 pathogenic) indicate that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no reported pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | 6-33438444-C-G | 1 | 6.20e-7 | -3.454 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.0 | -0.05 | Likely Benign | 0.16 | Likely Benign | 0.07 | Likely Benign | 0.273 | Likely Benign | -2.90 | Deleterious | 0.000 | Benign | 0.001 | Benign | -1.32 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.1048 | 0.4913 | -1 | 0 | 3.3 | 16.06 | |||||||||||||||||||||||||
| c.2198A>T | Q733L 2D AIThe SynGAP1 missense variant Q733L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -3.465 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -2.04 | Neutral | 0.905 | Possibly Damaging | 0.408 | Benign | 2.55 | Benign | 1.00 | Tolerated | 0.0736 | 0.4291 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2275A>C | M759L 2D AIThe SynGAP1 missense variant M759L is listed in ClinVar with an uncertain significance (ClinVar ID 942432.0) and is present in gnomAD (gnomAD ID 6‑33441740‑A‑C). All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign classifications. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign impact, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | Uncertain | 1 | 6-33441740-A-C | 2 | 1.24e-6 | -2.431 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.53 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.84 | Benign | 1.00 | Tolerated | 3.99 | 5 | 0.1308 | 0.4005 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||||
| c.2275A>T | M759L 2D AIThe SynGAP1 missense variant M759L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | -2.431 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.53 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.84 | Benign | 1.00 | Tolerated | 3.99 | 5 | 0.1308 | 0.4005 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.2320G>A | A774T 2D  AIThe SynGAP1 missense variant A774T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -3.238 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.46 | Neutral | 0.037 | Benign | 0.063 | Benign | 4.20 | Benign | 0.39 | Tolerated | 0.1436 | 0.7578 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2563C>A | L855I 2D  AIThe SynGAP1 missense variant L855I is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess functional impact all converge on a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool in the dataset indicates pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.485558 | Uncertain | 0.285 | 0.823 | 0.625 | -4.721 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.63 | Neutral | 0.004 | Benign | 0.008 | Benign | 4.08 | Benign | 0.35 | Tolerated | 0.0981 | 0.3893 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2590G>C | A864P 2D AIThe SynGAP1 missense variant A864P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.549308 | Disordered | 0.611966 | Binding | 0.269 | 0.788 | 0.250 | -3.665 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.12 | Neutral | 0.586 | Possibly Damaging | 0.211 | Benign | 2.60 | Benign | 0.09 | Tolerated | 0.1941 | 0.5746 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2695A>C | I899L 2D  AIThe SynGAP1 missense variant I899L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.443727 | Uncertain | 0.292 | 0.928 | 0.375 | -1.137 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.36 | Neutral | 0.220 | Benign | 0.078 | Benign | 2.74 | Benign | 0.04 | Affected | 0.0863 | 0.2840 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2830G>T | G944C 2D AIThe SynGAP1 missense variant G944C is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6‑33443382‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote of the four contributing tools) also indicates Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which remains unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.852408 | Binding | 0.360 | 0.923 | 0.750 | 6-33443382-G-T | 1 | 6.20e-7 | -9.121 | Likely Pathogenic | 0.093 | Likely Benign | Likely Benign | 0.426 | Likely Benign | -1.79 | Neutral | 0.975 | Probably Damaging | 0.848 | Possibly Damaging | 3.70 | Benign | 0.00 | Affected | 4.32 | 4 | 0.1399 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.2849G>T | G950V 2D AIThe SynGAP1 missense variant G950V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that G950V is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -7.796 | In-Between | 0.093 | Likely Benign | Likely Benign | 0.428 | Likely Benign | -0.91 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.26 | Pathogenic | 0.01 | Affected | 0.1392 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2903G>T | G968V 2D AIThe SynGAP1 missense variant G968V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -6.152 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.239 | Likely Benign | -1.08 | Neutral | 0.918 | Possibly Damaging | 0.525 | Possibly Damaging | 4.19 | Benign | 0.11 | Tolerated | 0.1252 | 0.4036 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>G | N1027S 2D  AIThe SynGAP1 missense variant N1027S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -2.443 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.35 | Neutral | 0.068 | Benign | 0.039 | Benign | 2.77 | Benign | 0.50 | Tolerated | 0.3540 | 0.6874 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3302C>T | P1101L 2D AIThe SynGAP1 missense variant P1101L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for P1101L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -4.335 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -2.19 | Neutral | 0.770 | Possibly Damaging | 0.255 | Benign | 4.27 | Benign | 0.04 | Affected | 0.2310 | 0.6050 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3338G>T | G1113V 2D AIThe SynGAP1 missense variant G1113V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -5.708 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.98 | Neutral | 0.827 | Possibly Damaging | 0.456 | Possibly Damaging | 2.53 | Benign | 0.11 | Tolerated | 0.1333 | 0.4224 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3385C>G | L1129V 2D AIThe SynGAP1 missense variant L1129V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | -3.595 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.262 | Likely Benign | -0.87 | Neutral | 0.393 | Benign | 0.187 | Benign | 5.46 | Benign | 0.00 | Affected | 0.1814 | 0.3804 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3442A>C | M1148L 2D AIThe SynGAP1 missense variant M1148L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and there is no conflict with ClinVar status because no ClinVar classification exists. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.774279 | Binding | 0.343 | 0.835 | 0.875 | -1.777 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -1.13 | Neutral | 0.016 | Benign | 0.016 | Benign | 2.62 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1641 | 0.4135 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.3442A>T | M1148L 2D AIThe SynGAP1 missense variant M1148L is listed in ClinVar (ID 1010061.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.774279 | Binding | 0.343 | 0.835 | 0.875 | Uncertain | 1 | -1.777 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -1.13 | Neutral | 0.016 | Benign | 0.016 | Benign | 2.62 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1641 | 0.4135 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||
| c.34A>G | S12G 2D  AIThe SynGAP1 missense variant S12G is reported in gnomAD (ID 6‑33420298‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect for S12G, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.490599 | Uncertain | 0.355 | 0.916 | 0.500 | 6-33420298-A-G | -4.229 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.079 | Likely Benign | 0.22 | Neutral | 0.103 | Benign | 0.015 | Benign | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2876 | 0.5080 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||
| c.3848C>T | P1283L 2D AIThe SynGAP1 missense variant P1283L is listed in ClinVar (ID 536994.0) as Benign and is present in gnomAD (gnomAD ID 6‑33447896‑C‑T). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic prediction. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts Benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, aligning with the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.819686 | Binding | 0.484 | 0.732 | 0.875 | Benign | 1 | 6-33447896-C-T | 32 | 2.06e-5 | -3.740 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -1.04 | Neutral | 0.005 | Benign | 0.003 | Benign | 2.76 | Benign | 0.06 | Tolerated | 3.77 | 5 | 0.1878 | 0.4716 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.3880G>A | A1294T 2D AIThe SynGAP1 missense variant A1294T is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447928‑G‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, five tools predict pathogenicity versus four predicting benign, and the lack of ClinVar evidence does not contradict these findings. Thus, the variant is most likely pathogenic based on the current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447928-G-A | -3.966 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.290 | Likely Benign | -3.04 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1244 | 0.5479 | 0 | 1 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||
| c.3898C>A | P1300T 2D  AIThe SynGAP1 missense variant P1300T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.885826 | Binding | 0.400 | 0.834 | 0.875 | -4.522 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -0.64 | Neutral | 0.649 | Possibly Damaging | 0.266 | Benign | 2.87 | Benign | 0.21 | Tolerated | 0.1546 | 0.4729 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3919C>A | P1307T 2D AIThe SynGAP1 missense variant P1307T is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly suggests that P1307T is most likely benign, and this conclusion is consistent with the lack of ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.913511 | Binding | 0.491 | 0.901 | 0.875 | -4.262 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.072 | Likely Benign | 0.20 | Neutral | 0.386 | Benign | 0.266 | Benign | 3.07 | Benign | 0.60 | Tolerated | 0.1732 | 0.5436 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3919C>T | P1307S 2D  AIThe SynGAP1 missense variant P1307S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.913511 | Binding | 0.491 | 0.901 | 0.875 | -3.700 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.049 | Likely Benign | 0.45 | Neutral | 0.239 | Benign | 0.157 | Benign | 3.09 | Benign | 0.48 | Tolerated | 0.3652 | 0.4846 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.955G>T | A319S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. No prediction or stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -7.109 | In-Between | 0.093 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.2 | 0.02 | Likely Benign | 0.09 | Likely Benign | 0.13 | Likely Benign | 0.165 | Likely Benign | -0.58 | Neutral | 0.978 | Probably Damaging | 0.754 | Possibly Damaging | 2.02 | Pathogenic | 0.12 | Tolerated | 0.2537 | 0.4703 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1109G>T | G370V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster into two groups: benign (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (FoldX, Rosetta, Foldetta, and FATHMM). High‑accuracy assessments further refine this picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. Overall, the majority of evidence points toward a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -5.328 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 4.98 | Destabilizing | 3.8 | 5.61 | Destabilizing | 5.30 | Destabilizing | -0.43 | Likely Benign | 0.427 | Likely Benign | 0.03 | Neutral | 0.000 | Benign | 0.000 | Benign | 1.32 | Pathogenic | 0.29 | Tolerated | 0.1306 | 0.4198 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2350G>T | A784S 2D AIThe SynGAP1 missense variant A784S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | -2.233 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.027 | Likely Benign | 0.41 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.72 | Benign | 0.67 | Tolerated | 0.2624 | 0.5171 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2413C>G | L805V 2D AIThe SynGAP1 missense variant L805V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign. Foldetta results are not available, so they do not influence the conclusion. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | -2.445 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -1.16 | Neutral | 0.003 | Benign | 0.008 | Benign | 2.65 | Benign | 0.00 | Affected | 0.1553 | 0.3493 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2561G>A | R854H 2D  AIThe SynGAP1 missense variant R854H is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443113‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.488780 | Uncertain | 0.277 | 0.815 | 0.750 | Uncertain | 1 | 6-33443113-G-A | 4 | 2.48e-6 | -3.686 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -1.38 | Neutral | 0.997 | Probably Damaging | 0.899 | Possibly Damaging | 4.07 | Benign | 0.04 | Affected | 3.88 | 3 | 0.3103 | 0.2202 | 2 | 0 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||
| c.2671C>G | L891V 2D AIThe SynGAP1 missense variant L891V is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.505861 | Binding | 0.305 | 0.923 | 0.750 | -4.992 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.65 | Neutral | 0.057 | Benign | 0.032 | Benign | 2.72 | Benign | 0.70 | Tolerated | 0.1614 | 0.2945 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.306G>C | L102F 2D  AIThe SynGAP1 missense variant L102F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.696014 | Binding | 0.357 | 0.885 | 0.625 | -4.712 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.80 | Neutral | 0.984 | Probably Damaging | 0.969 | Probably Damaging | 4.13 | Benign | 0.00 | Affected | 0.0863 | 0.3205 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.306G>T | L102F 2D AIThe SynGAP1 missense variant L102F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.696014 | Binding | 0.357 | 0.885 | 0.625 | -4.712 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -0.80 | Neutral | 0.984 | Probably Damaging | 0.969 | Probably Damaging | 4.13 | Benign | 0.00 | Affected | 0.0863 | 0.3205 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3155G>T | G1052V 2D AIThe SynGAP1 missense variant G1052V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign, while the high‑accuracy AlphaMissense‑Optimized score is benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign. In contrast, polyPhen‑2 HumDiv and HumVar both predict pathogenic, and ESM1b remains uncertain. No Foldetta stability assessment is available, so it does not influence the overall interpretation. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -7.717 | In-Between | 0.094 | Likely Benign | Likely Benign | 0.452 | Likely Benign | -0.12 | Neutral | 0.901 | Possibly Damaging | 0.619 | Possibly Damaging | 3.90 | Benign | 0.19 | Tolerated | 0.1329 | 0.3499 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3361A>T | S1121C 2D AIThe SynGAP1 missense variant S1121C is listed in gnomAD (ID 6‑33443913‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | 6-33443913-A-T | -7.431 | In-Between | 0.094 | Likely Benign | Likely Benign | 0.354 | Likely Benign | -0.99 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 5.43 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1705 | 0.5691 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3371G>T | G1124V 2D AIThe SynGAP1 missense variant G1124V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that G1124V is most likely benign, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | -6.980 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.315 | Likely Benign | -0.96 | Neutral | 0.586 | Possibly Damaging | 0.172 | Benign | 4.75 | Benign | 0.00 | Affected | 0.1299 | 0.3888 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3380G>T | G1127V 2D AIThe SynGAP1 missense variant G1127V is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443932‑G‑T). All available in silico predictors classify the change as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic prediction. Grouping by agreement, the benign‑predicting tools comprise the entire set, while no pathogenic predictions exist. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Uncertain | 1 | 6-33443932-G-T | 1 | 6.69e-7 | -6.097 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.230 | Likely Benign | -1.01 | Neutral | 0.004 | Benign | 0.005 | Benign | 4.81 | Benign | 0.17 | Tolerated | 4.32 | 4 | 0.1281 | 0.3669 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.3851T>A | L1284Q 2D  AIThe SynGAP1 missense variant L1284Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.812494 | Disordered | 0.824557 | Binding | 0.441 | 0.748 | 0.875 | -4.730 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.138 | Likely Benign | -2.80 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.48 | Pathogenic | 0.01 | Affected | 0.1065 | 0.0488 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3959C>T | P1320L 2D AIThe SynGAP1 missense variant P1320L is reported in gnomAD (variant ID 6‑33451833‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta’s protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect for P1320L, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.946297 | Binding | 0.510 | 0.833 | 0.750 | 6-33451833-C-T | -5.187 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -1.22 | Neutral | 0.994 | Probably Damaging | 0.981 | Probably Damaging | 4.18 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2622 | 0.6744 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||
| c.4028A>C | H1343P 2D  AIThe SynGAP1 missense variant H1343P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.983646 | Binding | 0.350 | 0.677 | 0.875 | -2.696 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -1.23 | Neutral | 0.659 | Possibly Damaging | 0.104 | Benign | 4.02 | Benign | 0.00 | Affected | 0.2333 | 0.4220 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.1153T>A | S385T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S385T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumVar and SIFT predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM‑Consensus, and Foldetta (combining FoldX‑MD and Rosetta outputs)—all indicate a benign effect. Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.733139 | Disordered | 0.425480 | Uncertain | 0.341 | 0.925 | 0.750 | -5.904 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.2 | 0.05 | Likely Benign | 0.35 | Likely Benign | -0.12 | Likely Benign | 0.214 | Likely Benign | -0.31 | Neutral | 0.140 | Benign | 0.481 | Possibly Damaging | 4.64 | Benign | 0.03 | Affected | 0.2266 | 0.6259 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1270G>A | V424I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V424I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. The only inconclusive results come from FoldX (uncertain) and Foldetta (uncertain). When high‑accuracy methods are considered separately, AlphaMissense‑Optimized predicts benign, the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also predicts benign, while Foldetta remains uncertain. No tool predicts pathogenicity. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -3.814 | Likely Benign | 0.095 | Likely Benign | Likely Benign | -1.04 | Ambiguous | 0.1 | -0.48 | Likely Benign | -0.76 | Ambiguous | 0.02 | Likely Benign | 0.084 | Likely Benign | -0.15 | Neutral | 0.013 | Benign | 0.006 | Benign | 3.50 | Benign | 0.63 | Tolerated | 0.0874 | 0.2609 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.1609G>T | A537S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A537S is reported in gnomAD (6-33438852-G-T) and has no ClinVar entry. Consensus from multiple in silico predictors indicates a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while polyPhen‑2 (HumDiv and HumVar) and FATHMM predict pathogenicity. When predictions are grouped, the majority of tools (ten) support benign, whereas three tools support pathogenic. High‑accuracy assessments further reinforce the benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Consequently, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.037313 | Uncertain | 0.928 | 0.362 | 0.000 | 6-33438852-G-T | 1 | 6.20e-7 | -3.602 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.28 | Likely Benign | 0.0 | 0.46 | Likely Benign | 0.37 | Likely Benign | 0.28 | Likely Benign | 0.206 | Likely Benign | -0.66 | Neutral | 0.528 | Possibly Damaging | 0.592 | Possibly Damaging | -1.25 | Pathogenic | 0.60 | Tolerated | 3.37 | 35 | 0.2729 | 0.4431 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||
| c.186T>A | D62E 2D  AIThe SynGAP1 missense variant D62E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -2.653 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -0.19 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.49 | Benign | 0.00 | Affected | 0.1884 | 0.5800 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.186T>G | D62E 2D AIThe SynGAP1 missense variant D62E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -2.653 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -0.19 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.49 | Benign | 0.00 | Affected | 0.1884 | 0.5800 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2032A>T | S678C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Tools with uncertain results are Rosetta and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (2 benign vs. 1 pathogenic, 1 uncertain), and Foldetta also predicts benign. No prediction or folding stability result is missing or inconclusive. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -7.879 | In-Between | 0.095 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.2 | 0.55 | Ambiguous | 0.38 | Likely Benign | 0.35 | Likely Benign | 0.094 | Likely Benign | -3.31 | Deleterious | 0.947 | Possibly Damaging | 0.527 | Possibly Damaging | 3.37 | Benign | 0.01 | Affected | 0.1080 | 0.5875 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.2201C>T | P734L 2D AIThe SynGAP1 missense variant P734L is reported in gnomAD (variant ID 6‑33441666‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | 6-33441666-C-T | 3 | 1.86e-6 | -3.472 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -2.11 | Neutral | 0.897 | Possibly Damaging | 0.330 | Benign | 2.69 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.2390 | 0.5059 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2263A>G | M755V 2D  AIThe SynGAP1 missense variant M755V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -3.804 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.80 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.73 | Benign | 0.27 | Tolerated | 0.2809 | 0.2873 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2578G>A | V860I 2D AIThe SynGAP1 missense variant V860I is listed in ClinVar (ID 411591) as Benign and is present in the gnomAD database (6‑33443130‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports Benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Benign, while a Foldetta stability analysis is unavailable. Overall, the majority of computational evidence points to a benign impact, which is consistent with the ClinVar designation and does not contradict the reported clinical status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.518121 | Binding | 0.269 | 0.803 | 0.250 | Benign/Likely benign | 2 | 6-33443130-G-A | 21 | 1.30e-5 | -4.516 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.42 | Neutral | 0.009 | Benign | 0.006 | Benign | 4.24 | Benign | 0.00 | Affected | 3.77 | 5 | 0.0842 | 0.4477 | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||||
| c.2807C>G | A936G 2D AIThe SynGAP1 missense variant A936G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.973218 | Binding | 0.319 | 0.874 | 0.625 | -2.720 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -1.34 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.48 | Pathogenic | 0.12 | Tolerated | 0.2380 | 0.4529 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2897A>T | H966L 2D AIThe SynGAP1 missense variant H966L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No pathogenic predictions are present among the evaluated algorithms. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign. Foldetta results are not available for this variant. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -6.119 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -1.74 | Neutral | 0.174 | Benign | 0.062 | Benign | 4.05 | Benign | 0.35 | Tolerated | 0.1514 | 0.5316 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2995T>C | S999P 2D  AIThe SynGAP1 missense variant S999P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for S999P, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -2.279 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -1.05 | Neutral | 0.966 | Probably Damaging | 0.773 | Possibly Damaging | 2.65 | Benign | 0.04 | Affected | 0.1963 | 0.6268 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3157A>T | S1053C 2D AIThe SynGAP1 missense variant S1053C is catalogued in gnomAD (ID 6‑33443709‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The ESM1b score is uncertain, providing no clear direction. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the majority of reliable predictors classify S1053C as benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | 6-33443709-A-T | -7.574 | In-Between | 0.095 | Likely Benign | Likely Benign | 0.220 | Likely Benign | -0.61 | Neutral | 0.977 | Probably Damaging | 0.777 | Possibly Damaging | 5.30 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.1675 | 0.5895 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3343A>T | I1115F 2D AIThe SynGAP1 missense variant I1115F is reported in gnomAD (ID 6‑33443895‑A‑T) and has no ClinVar entry. All available in silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are not available. Based on the unanimous benign predictions and the lack of a ClinVar pathogenic classification, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | 6-33443895-A-T | -3.426 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.98 | Neutral | 0.230 | Benign | 0.098 | Benign | 2.71 | Benign | 0.19 | Tolerated | 4.32 | 2 | 0.0769 | 0.4338 | 0 | 1 | -1.7 | 34.02 | ||||||||||||||||||||||||||||||||||||
| c.3374G>T | G1125V 2D AIThe SynGAP1 missense variant G1125V is reported in gnomAD (variant ID 6-33443926-G-T) but has no ClinVar entry. Functional prediction tools cluster into two groups: six tools (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score all predict a benign effect, whereas three tools (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT) predict pathogenicity. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443926-G-T | -6.776 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.320 | Likely Benign | -0.99 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.55 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1407 | 0.3688 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.3832C>A | P1278T 2D AIThe SynGAP1 missense variant P1278T is catalogued in gnomAD (ID 6‑33447880‑C‑A) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Overall, the computational evidence overwhelmingly supports a benign effect, and this assessment does not contradict any ClinVar classification. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.806955 | Binding | 0.532 | 0.722 | 0.750 | 6-33447880-C-A | -5.505 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -0.60 | Neutral | 0.059 | Benign | 0.026 | Benign | 2.73 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.1825 | 0.4053 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||
| c.56C>T | A19V 2D  AIThe SynGAP1 missense variant A19V is reported in gnomAD (ID 6‑33420320‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Overall, the consensus of available predictions points to a benign impact for A19V, and this conclusion is not contradicted by ClinVar status, which currently lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | 6-33420320-C-T | -3.157 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.063 | Likely Benign | 0.42 | Neutral | 0.371 | Benign | 0.036 | Benign | 4.27 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1804 | 0.6942 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||
| c.902C>T | A301V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A301V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenicity. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Thus, the overall evidence supports a benign classification for A301V, and this conclusion is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.258424 | Uncertain | 0.647 | 0.272 | 0.375 | -2.476 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.25 | Likely Benign | 0.1 | 0.48 | Likely Benign | 0.37 | Likely Benign | 0.06 | Likely Benign | 0.116 | Likely Benign | -0.49 | Neutral | 0.997 | Probably Damaging | 0.983 | Probably Damaging | 4.14 | Benign | 0.26 | Tolerated | 0.1096 | 0.6264 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1124G>C | G375A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G375A is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact. Overall, the majority of evidence points to a benign effect; this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -5.986 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 2.52 | Destabilizing | 1.0 | 3.16 | Destabilizing | 2.84 | Destabilizing | -0.09 | Likely Benign | 0.419 | Likely Benign | -0.61 | Neutral | 0.020 | Benign | 0.008 | Benign | 1.33 | Pathogenic | 0.27 | Tolerated | 0.3991 | 0.5242 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1171G>A | G391S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and FoldX. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also as likely benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact for G391S, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -5.531 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 2.14 | Destabilizing | 0.6 | 1.01 | Ambiguous | 1.58 | Ambiguous | 0.06 | Likely Benign | 0.462 | Likely Benign | -0.54 | Neutral | 0.978 | Probably Damaging | 0.777 | Possibly Damaging | 1.33 | Pathogenic | 0.35 | Tolerated | 0.2820 | 0.5097 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1189T>G | C397G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach consensus all indicate a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies it as “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | 1.244 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.68 | Ambiguous | 0.2 | 1.42 | Ambiguous | 1.05 | Ambiguous | 0.04 | Likely Benign | 0.272 | Likely Benign | 1.51 | Neutral | 0.276 | Benign | 0.066 | Benign | 5.93 | Benign | 0.60 | Tolerated | 0.3678 | 0.3729 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.120T>A | D40E 2D AIThe SynGAP1 missense variant D40E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.520 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -0.21 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2764 | 0.8141 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.120T>G | D40E 2D AIThe SynGAP1 missense variant D40E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this assessment does not contradict the ClinVar record, which contains no pathogenic claim. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.520 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -0.21 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2764 | 0.8141 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2239G>C | V747L 2D  AIThe SynGAP1 missense variant V747L (ClinVar ID 1985039.0) is listed as ClinVar status Uncertain and is present in gnomAD (6‑33441704‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of computational evidence supports a benign classification, which is consistent with the ClinVar Uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | Uncertain | 1 | 6-33441704-G-C | 2 | 1.24e-6 | -2.790 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.52 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.67 | Benign | 0.00 | Affected | 4.32 | 2 | 0.0699 | 0.3674 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||
| c.2239G>T | V747L 2D AIThe SynGAP1 missense variant V747L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -2.790 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.52 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.67 | Benign | 0.00 | Affected | 4.32 | 2 | 0.0699 | 0.3674 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.2368A>G | T790A 2D  AIThe SynGAP1 T790A missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, while the SGM Consensus remains inconclusive and Foldetta data are unavailable. Overall, the majority of standard predictors indicate pathogenicity, but the single high‑accuracy tool that is available suggests a benign effect, and no high‑accuracy tool provides a definitive pathogenic verdict. Consequently, the variant is most likely pathogenic according to the bulk of predictions, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -4.337 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.64 | Deleterious | 0.992 | Probably Damaging | 0.989 | Probably Damaging | 2.35 | Pathogenic | 0.02 | Affected | 0.4157 | 0.4207 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2557G>T | G853C 2D AIThe SynGAP1 G853C missense variant has no ClinVar record and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.496246 | Uncertain | 0.284 | 0.815 | 0.625 | -7.021 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.120 | Likely Benign | -1.65 | Neutral | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.1292 | 0.4944 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2567A>C | N856T 2D  AIThe SynGAP1 missense variant N856T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; no Foldetta stability result is available, so it does not influence the overall interpretation. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.477615 | Uncertain | 0.263 | 0.827 | 0.500 | -3.046 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.037 | Likely Benign | -1.63 | Neutral | 0.818 | Possibly Damaging | 0.559 | Possibly Damaging | 4.13 | Benign | 0.17 | Tolerated | 0.1493 | 0.8096 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.2581T>A | S861T 2D AIThe SynGAP1 missense variant S861T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), also reports a likely benign outcome. AlphaMissense‑Optimized likewise predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.540903 | Binding | 0.285 | 0.797 | 0.250 | -4.462 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.022 | Likely Benign | -0.83 | Neutral | 0.043 | Benign | 0.026 | Benign | 3.99 | Benign | 0.14 | Tolerated | 0.1262 | 0.6245 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2881C>A | H961N 2D  AIThe SynGAP1 missense variant H961N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H961N is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.989835 | Disordered | 0.984562 | Binding | 0.323 | 0.893 | 0.750 | -8.561 | Likely Pathogenic | 0.096 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -0.32 | Neutral | 0.069 | Benign | 0.036 | Benign | 4.17 | Benign | 0.81 | Tolerated | 0.2074 | 0.3695 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3311C>T | P1104L 2D AIThe SynGAP1 missense variant P1104L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also predicts Likely Benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, points to a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | -3.846 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.33 | Neutral | 0.626 | Possibly Damaging | 0.168 | Benign | 2.81 | Benign | 1.00 | Tolerated | 0.2264 | 0.6795 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3328A>T | S1110C 2D AIThe SynGAP1 missense variant S1110C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign (2 benign vs. 1 pathogenic vote). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy consensus—points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -7.250 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -2.12 | Neutral | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 2.16 | Pathogenic | 0.01 | Affected | 0.1214 | 0.5954 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3352A>T | S1118C 2D AIThe SynGAP1 missense variant S1118C is listed in gnomAD (ID 6‑33443904‑A‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | 6-33443904-A-T | -7.402 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.311 | Likely Benign | -1.05 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 5.15 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1648 | 0.5695 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3399C>G | I1133M 2D  AIThe SynGAP1 missense variant I1133M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -3.409 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.223 | Likely Benign | -0.21 | Neutral | 0.016 | Benign | 0.009 | Benign | 5.45 | Benign | 0.06 | Tolerated | 0.0764 | 0.3685 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3769T>G | S1257A 2D AIThe SynGAP1 missense variant S1257A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus also reports a benign likelihood. Foldetta results are unavailable, so they do not influence the conclusion. Overall, the preponderance of evidence points to a benign impact for S1257A, and this assessment is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.482380 | Uncertain | 0.889 | 0.572 | 0.375 | -3.937 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -1.02 | Neutral | 0.454 | Possibly Damaging | 0.266 | Benign | 2.64 | Benign | 0.26 | Tolerated | 0.4083 | 0.3800 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.3790G>A | G1264S 2D AIThe SynGAP1 missense variant G1264S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: benign predictions are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized classifies the variant as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Foldetta results are unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.429200 | Structured | 0.762385 | Binding | 0.897 | 0.579 | 0.000 | -0.797 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.147 | Likely Benign | 0.82 | Neutral | 0.062 | Benign | 0.033 | Benign | 2.94 | Benign | 1.00 | Tolerated | 0.2376 | 0.4256 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||||||||
| c.3865A>C | K1289Q 2D  AIThe SynGAP1 missense variant K1289Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.828700 | Binding | 0.548 | 0.787 | 0.625 | -2.940 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.066 | Likely Benign | 1.55 | Neutral | 0.004 | Benign | 0.004 | Benign | 3.09 | Benign | 1.00 | Tolerated | 0.4002 | 0.0590 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3893A>C | Q1298P 2D  AIThe SynGAP1 missense variant Q1298P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.895297 | Binding | 0.410 | 0.821 | 0.750 | -1.819 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.196 | Likely Benign | -2.93 | Deleterious | 0.586 | Possibly Damaging | 0.105 | Benign | 2.76 | Benign | 0.03 | Affected | 0.2237 | 0.4025 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3940C>A | P1314T 2D AIThe SynGAP1 missense variant P1314T is present in gnomAD (ID 6‑33451814‑C‑A) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect. Benign predictions come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Overall, the computational evidence overwhelmingly supports a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this is consistent with the lack of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.971592 | Binding | 0.467 | 0.903 | 0.750 | 6-33451814-C-A | -4.083 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.066 | Likely Benign | 1.26 | Neutral | 0.061 | Benign | 0.045 | Benign | 4.32 | Benign | 0.97 | Tolerated | 3.77 | 5 | 0.1791 | 0.5626 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||
| c.3962C>T | P1321L 2D AIThe SynGAP1 missense variant P1321L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.933505 | Binding | 0.463 | 0.828 | 0.875 | -4.892 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -0.81 | Neutral | 0.115 | Benign | 0.009 | Benign | 4.28 | Benign | 0.08 | Tolerated | 0.2626 | 0.6222 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.40C>G | P14A 2D AIThe SynGAP1 missense variant P14A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign status: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta results are unavailable. Overall, the majority of computational evidence indicates that P14A is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.433034 | Structured | 0.471596 | Uncertain | 0.399 | 0.909 | 0.375 | -2.919 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.01 | Neutral | 0.100 | Benign | 0.007 | Benign | 4.24 | Benign | 0.00 | Affected | 0.3928 | 0.5543 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.1181A>G | K394R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K394R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. Only SIFT predicts a pathogenic outcome, while Rosetta and Foldetta are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, and Foldetta remains uncertain. Overall, the preponderance of evidence supports a benign classification for K394R, and this conclusion does not contradict the absence of a ClinVar report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -4.902 | Likely Benign | 0.097 | Likely Benign | Likely Benign | -0.01 | Likely Benign | 0.1 | 1.19 | Ambiguous | 0.59 | Ambiguous | 0.42 | Likely Benign | 0.335 | Likely Benign | -1.97 | Neutral | 0.141 | Benign | 0.091 | Benign | 5.11 | Benign | 0.03 | Affected | 0.5488 | 0.2075 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||
| c.1609G>A | A537T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A537T is catalogued in gnomAD (6‑33438852‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign; the SGM‑Consensus (majority vote) is Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts Benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.037313 | Uncertain | 0.928 | 0.362 | 0.000 | 6-33438852-G-A | 2 | 1.24e-6 | -4.704 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.35 | Likely Benign | 0.0 | 0.29 | Likely Benign | 0.32 | Likely Benign | 0.37 | Likely Benign | 0.210 | Likely Benign | -1.41 | Neutral | 0.953 | Possibly Damaging | 0.602 | Possibly Damaging | -1.27 | Pathogenic | 0.44 | Tolerated | 3.37 | 35 | 0.1520 | 0.5440 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||
| c.17C>G | A6G 2D AIThe SynGAP1 missense variant A6G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.566480 | Disordered | 0.549054 | Binding | 0.377 | 0.920 | 0.875 | -2.786 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.107 | Likely Benign | 0.31 | Neutral | 0.052 | Benign | 0.004 | Benign | 4.08 | Benign | 0.00 | Affected | 0.2139 | 0.4369 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2014A>T | T672S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T672S is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign or likely benign. No tool predicts pathogenicity. High‑accuracy methods corroborate this: AlphaMissense‑Optimized reports benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | -4.932 | Likely Benign | 0.097 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.1 | 0.29 | Likely Benign | 0.08 | Likely Benign | 0.61 | Ambiguous | 0.044 | Likely Benign | -2.32 | Neutral | 0.006 | Benign | 0.010 | Benign | 3.43 | Benign | 0.09 | Tolerated | 0.3005 | 0.4282 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2122C>G | L708V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results are from FoldX (uncertain) and Foldetta (uncertain). High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, while Foldetta remains uncertain. Overall, the evidence strongly indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -4.361 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 1.59 | Ambiguous | 0.0 | 0.12 | Likely Benign | 0.86 | Ambiguous | -0.02 | Likely Benign | 0.122 | Likely Benign | -0.23 | Neutral | 0.158 | Benign | 0.035 | Benign | 3.35 | Benign | 0.94 | Tolerated | 0.1094 | 0.2460 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2258C>T | A753V 2D AIThe SynGAP1 missense variant A753V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of computational evidence indicates that A753V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -3.759 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.55 | Neutral | 0.669 | Possibly Damaging | 0.192 | Benign | 2.71 | Benign | 0.18 | Tolerated | 0.1344 | 0.5953 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2333A>G | N778S 2D AIThe SynGAP1 missense variant N778S is reported in gnomAD (ID 6‑33442491‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and HumVar—predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | 6-33442491-A-G | 1 | 1.28e-6 | -2.711 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -0.61 | Neutral | 0.843 | Possibly Damaging | 0.893 | Possibly Damaging | 4.32 | Benign | 0.86 | Tolerated | 3.64 | 6 | 0.4285 | 0.6890 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||||||||||
| c.2408A>G | K803R 2D AIThe SynGAP1 missense variant K803R is listed in ClinVar (ID 834618.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—SIFT and FATHMM—predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | Uncertain | 1 | -2.281 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.018 | Likely Benign | -1.52 | Neutral | 0.103 | Benign | 0.038 | Benign | 2.38 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.4857 | 0.1715 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||
| c.2629C>G | L877V 2D AIThe SynGAP1 missense variant L877V is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.653063 | Disordered | 0.634010 | Binding | 0.265 | 0.875 | 0.250 | -5.063 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.022 | Likely Benign | -0.10 | Neutral | 0.232 | Benign | 0.109 | Benign | 2.71 | Benign | 0.26 | Tolerated | 0.1726 | 0.3178 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2701G>T | A901S 2D AIThe SynGAP1 missense variant A901S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess pathogenicity uniformly predict a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool in the dataset predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.489838 | Uncertain | 0.306 | 0.917 | 0.375 | -3.284 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.62 | Neutral | 0.009 | Benign | 0.015 | Benign | 2.73 | Benign | 0.35 | Tolerated | 0.2695 | 0.6582 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2734A>G | T912A 2D AIThe SynGAP1 missense variant T912A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.740671 | Binding | 0.285 | 0.909 | 0.250 | -3.084 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.50 | Neutral | 0.973 | Probably Damaging | 0.856 | Possibly Damaging | 4.03 | Benign | 0.00 | Affected | 0.3754 | 0.3841 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2818G>T | G940C 2D AIThe SynGAP1 missense variant G940C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this conclusion, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.920635 | Binding | 0.383 | 0.902 | 0.625 | -8.158 | Likely Pathogenic | 0.097 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.40 | Neutral | 0.996 | Probably Damaging | 0.905 | Possibly Damaging | 2.70 | Benign | 0.11 | Tolerated | 0.1307 | 0.4354 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2866T>A | S956T 2D AIThe SynGAP1 missense variant S956T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for S956T, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -5.404 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.45 | Neutral | 0.369 | Benign | 0.159 | Benign | 2.00 | Pathogenic | 0.08 | Tolerated | 0.2244 | 0.5727 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2872C>A | H958N 2D AIThe SynGAP1 missense variant H958N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.976011 | Binding | 0.371 | 0.913 | 0.750 | -8.644 | Likely Pathogenic | 0.097 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.56 | Neutral | 0.836 | Possibly Damaging | 0.232 | Benign | 4.17 | Benign | 1.00 | Tolerated | 0.2358 | 0.3638 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2918G>T | G973V 2D AIThe SynGAP1 missense variant G973V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the single pathogenic prediction. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -4.688 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -0.76 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1392 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3032G>C | G1011A 2D AIThe SynGAP1 missense variant G1011A is catalogued in gnomAD (ID 6‑33443584‑G‑C) and has no ClinVar entry. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic outcome. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | 6-33443584-G-C | -4.349 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.04 | Neutral | 0.139 | Benign | 0.089 | Benign | 2.89 | Benign | 0.64 | Tolerated | 3.77 | 5 | 0.3641 | 0.4571 | 0 | 1 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||||
| c.3167G>T | G1056V 2D AIThe SynGAP1 missense variant G1056V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence (7 benign vs 2 pathogenic) supports a benign classification. This consensus does not contradict ClinVar status, which has no entry for this variant. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -8.130 | Likely Pathogenic | 0.097 | Likely Benign | Likely Benign | 0.448 | Likely Benign | -0.24 | Neutral | 0.292 | Benign | 0.110 | Benign | 1.83 | Pathogenic | 0.08 | Tolerated | 0.1488 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3289C>A | P1097T 2D AIThe SynGAP1 missense variant P1097T is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the computational evidence strongly supports a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | -5.000 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -1.38 | Neutral | 0.009 | Benign | 0.013 | Benign | 2.61 | Benign | 0.21 | Tolerated | 0.1751 | 0.6567 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3737A>G | K1246R 2D AIThe SynGAP1 missense variant K1246R is reported in gnomAD (ID 6‑33446729‑A‑G) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.750527 | Disordered | 0.375382 | Uncertain | 0.887 | 0.564 | 0.625 | 6-33446729-A-G | 1 | 6.20e-7 | -2.444 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.86 | Neutral | 0.031 | Benign | 0.017 | Benign | 2.66 | Benign | 0.04 | Affected | 3.77 | 5 | 0.3790 | 0.0515 | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||
| c.3845A>C | E1282A 2D  AIThe SynGAP1 missense variant E1282A is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No pathogenic predictions are present. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the computational evidence overwhelmingly supports a benign impact, and this conclusion does not contradict any ClinVar status, as none exists for E1282A. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.817364 | Binding | 0.465 | 0.725 | 0.875 | -1.980 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.118 | Likely Benign | 0.52 | Neutral | 0.026 | Benign | 0.018 | Benign | 2.94 | Benign | 0.38 | Tolerated | 0.3411 | 0.5358 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3935C>G | A1312G 2D AIThe SynGAP1 missense variant A1312G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.767246 | Disordered | 0.971112 | Binding | 0.392 | 0.902 | 0.750 | -3.735 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -2.25 | Neutral | 0.978 | Probably Damaging | 0.983 | Probably Damaging | 3.15 | Benign | 0.00 | Affected | 0.2241 | 0.5058 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3938C>T | P1313L 2D AIThe SynGAP1 missense variant P1313L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.970301 | Binding | 0.452 | 0.902 | 0.750 | -4.588 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.082 | Likely Benign | 1.90 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.30 | Benign | 1.00 | Tolerated | 0.2489 | 0.6948 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.43G>C | A15P 2D AIThe SynGAP1 missense variant A15P is listed in ClinVar (ID 3688743.0) with an *Uncertain* clinical significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as *Likely Benign*. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.466055 | Uncertain | 0.330 | 0.912 | 0.375 | Uncertain | 1 | -3.436 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.23 | Neutral | 0.880 | Possibly Damaging | 0.123 | Benign | 4.09 | Benign | 0.00 | Affected | 0.2573 | 0.6988 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||
| c.1720C>A | L574M 2D  AIThe SynGAP1 missense variant L574M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. No predictions or stability results are missing or inconclusive. Based on the overall consensus, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | -7.195 | In-Between | 0.098 | Likely Benign | Likely Benign | 0.14 | Likely Benign | 0.2 | 0.34 | Likely Benign | 0.24 | Likely Benign | -0.09 | Likely Benign | 0.113 | Likely Benign | 0.85 | Neutral | 0.691 | Possibly Damaging | 0.278 | Benign | -1.29 | Pathogenic | 0.11 | Tolerated | 0.0894 | 0.3087 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.2494C>G | Q832E 2D  AIThe SynGAP1 missense variant Q832E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, but this is the sole discordant call. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.619913 | Binding | 0.290 | 0.877 | 0.375 | -3.024 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.37 | Neutral | 0.652 | Possibly Damaging | 0.311 | Benign | 2.77 | Benign | 0.06 | Tolerated | 0.1262 | 0.1897 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2698A>T | T900S 2D AIThe SynGAP1 missense variant T900S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.465347 | Uncertain | 0.289 | 0.924 | 0.375 | -2.031 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.17 | Neutral | 0.224 | Benign | 0.171 | Benign | 2.70 | Benign | 0.71 | Tolerated | 0.3311 | 0.4301 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2815C>T | P939S 2D AIThe SynGAP1 missense variant P939S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) indicate that P939S is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.331 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -3.44 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.24 | Pathogenic | 0.00 | Affected | 0.3372 | 0.4517 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2822C>A | P941H 2D  AIThe SynGAP1 missense variant P941H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, two tools—polyPhen‑2 HumDiv and SIFT—classify the change as pathogenic. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; no Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.900790 | Binding | 0.403 | 0.906 | 0.625 | -4.439 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.09 | Neutral | 0.589 | Possibly Damaging | 0.309 | Benign | 2.71 | Benign | 0.00 | Affected | 0.1621 | 0.4826 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2884C>A | H962N 2D AIThe SynGAP1 missense variant H962N is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.984483 | Binding | 0.369 | 0.886 | 0.750 | -8.481 | Likely Pathogenic | 0.098 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.61 | Neutral | 0.174 | Benign | 0.045 | Benign | 4.18 | Benign | 0.24 | Tolerated | 0.1957 | 0.3216 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2899C>G | R967G 2D  AIThe SynGAP1 missense variant R967G is reported in gnomAD (ID 6‑33443451‑C‑G) and has no ClinVar entry. All evaluated in silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Based on the unanimous benign predictions and absence of a ClinVar pathogenic claim, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | 6-33443451-C-G | 1 | 6.20e-7 | -2.214 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.279 | Likely Benign | -0.93 | Neutral | 0.005 | Benign | 0.007 | Benign | 4.17 | Benign | 0.18 | Tolerated | 4.32 | 2 | 0.3187 | 0.4070 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.2972G>T | G991V 2D AIThe SynGAP1 missense variant G991V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.911393 | Binding | 0.286 | 0.920 | 0.750 | -4.129 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -1.61 | Neutral | 0.440 | Benign | 0.253 | Benign | 4.16 | Benign | 0.01 | Affected | 0.1148 | 0.3961 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2978C>T | P993L 2D AIThe SynGAP1 missense variant P993L is reported in ClinVar as “Not listed” and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -3.581 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -1.37 | Neutral | 0.224 | Benign | 0.138 | Benign | 4.14 | Benign | 0.01 | Affected | 0.2299 | 0.6697 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2998A>G | I1000V 2D  AIThe SynGAP1 missense variant I1000V is listed in ClinVar (ID 2572013.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools that assess evolutionary conservation and structural impact (REVEL, SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default) all converge on a benign outcome. No tool in the dataset predicts pathogenicity. High‑accuracy predictors reinforce this consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | Uncertain | 2 | -4.102 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -0.20 | Neutral | 0.437 | Benign | 0.170 | Benign | 2.76 | Benign | 0.81 | Tolerated | 4.32 | 4 | 0.1220 | 0.3404 | 3 | 4 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||
| c.3400A>C | T1134P 2D  AIThe SynGAP1 missense variant T1134P is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -2.993 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -0.57 | Neutral | 0.013 | Benign | 0.022 | Benign | 5.41 | Benign | 0.07 | Tolerated | 0.2001 | 0.5165 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.364C>T | P122S 2D AIThe SynGAP1 missense variant P122S is catalogued in gnomAD (ID 6‑33432229‑C‑T) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.672358 | Binding | 0.400 | 0.887 | 0.750 | 6-33432229-C-T | 1 | 6.20e-7 | -3.389 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.13 | Neutral | 0.036 | Benign | 0.025 | Benign | 4.22 | Benign | 0.31 | Tolerated | 3.61 | 5 | 0.4009 | 0.4759 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.3670C>A | L1224M 2D AIThe SynGAP1 missense variant L1224M is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign) all indicate a tolerated change. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. When focusing on high‑accuracy predictors, AlphaMissense‑Optimized remains benign and the SGM‑Consensus also supports a benign outcome; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -5.614 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.55 | Neutral | 0.994 | Probably Damaging | 0.925 | Probably Damaging | 2.38 | Pathogenic | 0.18 | Tolerated | 0.0660 | 0.2486 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3716C>T | A1239V 2D AIThe A1239V missense change occurs in a coiled‑coil region of SynGAP1. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. ClinVar has no entry for this variant, and it is not present in gnomAD. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -5.914 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -2.28 | Neutral | 0.425 | Benign | 0.233 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.0771 | 0.4518 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||||
| c.3832C>T | P1278S 2D AIThe SynGAP1 missense variant P1278S is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.806955 | Binding | 0.532 | 0.722 | 0.750 | -4.383 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.077 | Likely Benign | 0.28 | Neutral | 0.004 | Benign | 0.003 | Benign | 2.96 | Benign | 0.35 | Tolerated | 0.3616 | 0.3697 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.79C>T | P27S 2D AIThe SynGAP1 missense variant P27S is reported in gnomAD (ID 6‑33423488‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, and the SGM‑Consensus (derived from the majority of these high‑accuracy predictors) also reports a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign. Foldetta stability analysis is unavailable. Overall, the preponderance of evidence indicates that P27S is most likely benign, and this assessment does not contradict any ClinVar classification, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.437871 | Uncertain | 0.430 | 0.881 | 0.375 | 6-33423488-C-T | 1 | 6.20e-7 | -2.891 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -2.01 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 3.91 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3916 | 0.5443 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.955G>A | A319T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict a pathogenic effect are polyPhen2_HumDiv and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of reliable predictors classify the variant as benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -7.841 | In-Between | 0.098 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.3 | 0.70 | Ambiguous | 0.63 | Ambiguous | 0.30 | Likely Benign | 0.116 | Likely Benign | -1.35 | Neutral | 0.775 | Possibly Damaging | 0.306 | Benign | 1.92 | Pathogenic | 0.09 | Tolerated | 0.1274 | 0.6083 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||
| c.1019C>G | A340G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A340G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictors indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -3.763 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.66 | Ambiguous | 0.2 | 1.44 | Ambiguous | 1.05 | Ambiguous | 0.50 | Likely Benign | 0.044 | Likely Benign | -0.34 | Neutral | 0.267 | Benign | 0.127 | Benign | 1.92 | Pathogenic | 0.42 | Tolerated | 0.2355 | 0.4470 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1111A>T | S371C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the majority of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -6.330 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.19 | Likely Benign | 0.2 | -0.34 | Likely Benign | -0.08 | Likely Benign | 0.23 | Likely Benign | 0.450 | Likely Benign | -1.41 | Neutral | 0.875 | Possibly Damaging | 0.359 | Benign | 4.61 | Benign | 0.02 | Affected | 0.1786 | 0.6580 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.1118G>C | G373A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G373A is not reported in ClinVar and is absent from gnomAD. Functional prediction consensus shows a predominance of benign calls: REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all predict a benign effect. Pathogenic predictions are limited to SIFT and FoldX, while Rosetta and Foldetta yield uncertain results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta remains inconclusive. Overall, the majority of evidence indicates that G373A is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | -5.181 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 2.44 | Destabilizing | 0.8 | 0.69 | Ambiguous | 1.57 | Ambiguous | -0.01 | Likely Benign | 0.227 | Likely Benign | -0.47 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.93 | Benign | 0.01 | Affected | 0.4172 | 0.5053 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1127G>C | G376A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G376A missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. FoldX and Rosetta give uncertain results and are not considered evidence for either side. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -6.016 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 1.74 | Ambiguous | 0.3 | -0.84 | Ambiguous | 0.45 | Likely Benign | 0.00 | Likely Benign | 0.392 | Likely Benign | -0.44 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.33 | Pathogenic | 0.03 | Affected | 0.3868 | 0.4465 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.2017C>A | L673I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The remaining tools (FoldX, Rosetta, Foldetta, ESM1b) return uncertain or inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Taken together, the majority of reliable predictors indicate a benign effect, and there is no conflict with ClinVar status (which has no entry). Therefore, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -7.823 | In-Between | 0.099 | Likely Benign | Likely Benign | 1.51 | Ambiguous | 0.2 | 1.89 | Ambiguous | 1.70 | Ambiguous | -0.02 | Likely Benign | 0.036 | Likely Benign | -0.14 | Neutral | 0.535 | Possibly Damaging | 0.112 | Benign | 3.37 | Benign | 0.47 | Tolerated | 0.0910 | 0.3689 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2017C>G | L673V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Benign, and Foldetta as pathogenic, yielding one pathogenic versus two benign predictions. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -8.132 | Likely Pathogenic | 0.099 | Likely Benign | Likely Benign | 2.18 | Destabilizing | 0.3 | 2.10 | Destabilizing | 2.14 | Destabilizing | 0.10 | Likely Benign | 0.017 | Likely Benign | -0.58 | Neutral | 0.016 | Benign | 0.003 | Benign | 3.36 | Benign | 0.28 | Tolerated | 0.1448 | 0.3777 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2020A>G | T674A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly favor a benign effect: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, REVEL, FoldX, premPS, Foldetta, and the SGM‑Consensus score all indicate benign or likely benign. No tool predicts pathogenicity; the only inconclusive result comes from Rosetta, which is listed as uncertain. High‑accuracy methods corroborate the benign assessment: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the aggregate predictions, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -5.915 | Likely Benign | 0.099 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.0 | 0.89 | Ambiguous | 0.38 | Likely Benign | 0.29 | Likely Benign | 0.052 | Likely Benign | -1.48 | Neutral | 0.398 | Benign | 0.045 | Benign | 3.45 | Benign | 0.28 | Tolerated | 0.4042 | 0.4845 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.2228C>A | P743H 2D AIThe SynGAP1 missense variant P743H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of computational evidence points to a benign effect for P743H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | -5.649 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -1.99 | Neutral | 0.989 | Probably Damaging | 0.870 | Possibly Damaging | 2.70 | Benign | 0.01 | Affected | 0.1653 | 0.3762 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2230C>A | Q744K 2D 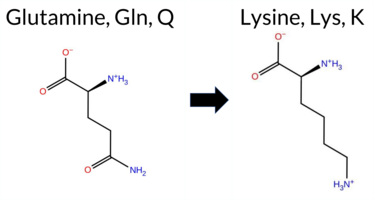 AIThe SynGAP1 missense variant Q744K is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -3.929 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.22 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.79 | Benign | 0.07 | Tolerated | 0.1784 | 0.3757 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2321C>G | A774G 2D AIThe SynGAP1 missense variant A774G is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -3.129 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.68 | Neutral | 0.135 | Benign | 0.152 | Benign | 4.16 | Benign | 0.22 | Tolerated | 0.2392 | 0.5436 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2666G>C | G889A 2D AIThe SynGAP1 missense variant G889A is listed in gnomAD (6‑33443218‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign or likely benign. Only FATHMM predicts a pathogenic outcome, representing a single dissenting signal. High‑accuracy assessments confirm the benign consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus likewise indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence supports a benign classification, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | 6-33443218-G-C | 1 | 6.20e-7 | -4.580 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.49 | Neutral | 0.245 | Benign | 0.096 | Benign | 2.47 | Pathogenic | 0.76 | Tolerated | 4.32 | 4 | 0.3838 | 0.4902 | 0 | 1 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||
| c.2671C>A | L891I 2D AIThe SynGAP1 missense variant L891I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.505861 | Binding | 0.305 | 0.923 | 0.750 | -5.803 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.71 | Neutral | 0.481 | Possibly Damaging | 0.202 | Benign | 2.74 | Benign | 0.14 | Tolerated | 0.0957 | 0.3494 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2704G>T | A902S 2D AIThe SynGAP1 missense variant A902S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for A902S, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.517703 | Binding | 0.319 | 0.919 | 0.375 | -4.176 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.74 | Neutral | 0.798 | Possibly Damaging | 0.433 | Benign | 2.63 | Benign | 0.27 | Tolerated | 0.2515 | 0.5565 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2815C>A | P939T 2D AIThe SynGAP1 missense variant P939T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while the SGM Consensus remains inconclusive and Foldetta is unavailable. Overall, more tools (five) predict pathogenicity than benign (four), suggesting the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -5.739 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -3.34 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.1788 | 0.5310 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||||||
| c.2887C>A | H963N 2D AIThe SynGAP1 missense variant H963N is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.983973 | Binding | 0.325 | 0.886 | 0.750 | -8.274 | Likely Pathogenic | 0.099 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.21 | Neutral | 0.369 | Benign | 0.120 | Benign | 4.18 | Benign | 0.16 | Tolerated | 0.2094 | 0.3596 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3146C>T | P1049L 2D AIThe SynGAP1 missense variant P1049L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -4.819 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -2.37 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.71 | Benign | 0.02 | Affected | 0.2267 | 0.5838 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3191A>T | Q1064L 2D AIThe SynGAP1 missense variant Q1064L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -3.492 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -1.16 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.20 | Benign | 0.13 | Tolerated | 0.1817 | 0.5485 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3356G>T | G1119V 2D AIThe SynGAP1 missense variant G1119V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | -6.428 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.352 | Likely Benign | -0.94 | Neutral | 0.918 | Possibly Damaging | 0.604 | Possibly Damaging | 3.91 | Benign | 0.31 | Tolerated | 0.1493 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.362C>G | A121G 2D AIThe SynGAP1 missense variant A121G is catalogued in gnomAD (ID 6‑33432227‑C‑G) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool examined—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No pathogenic predictions are reported. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Overall, the evidence strongly supports a benign effect for A121G, and this conclusion does not contradict any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.661304 | Binding | 0.362 | 0.888 | 0.750 | 6-33432227-C-G | 4 | 2.48e-6 | -3.123 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -0.52 | Neutral | 0.118 | Benign | 0.026 | Benign | 4.06 | Benign | 0.07 | Tolerated | 3.61 | 5 | 0.2407 | 0.5271 | 0 | 1 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.3703A>G | M1235V 2D AIThe SynGAP1 missense variant M1235V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. Only SIFT predicts a damaging effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this residue. Taken together, the preponderance of evidence indicates that M1235V is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.690604 | Disordered | 0.577958 | Binding | 0.872 | 0.532 | 0.125 | -3.870 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.54 | Neutral | 0.139 | Benign | 0.038 | Benign | 2.69 | Benign | 0.02 | Affected | 0.3048 | 0.3120 | 2 | 1 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||||||||||
| c.3902C>T | P1301L 2D AIThe SynGAP1 missense variant P1301L is listed in ClinVar (ID 4749342) with an uncertain significance annotation and is present in gnomAD (variant ID 6‑33451776‑C‑T). Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign effects. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this benign profile: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. A protein‑folding stability analysis with Foldetta is unavailable, so it does not influence the overall assessment. Overall, the computational evidence strongly supports a benign classification, which is consistent with the ClinVar uncertain status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.885064 | Binding | 0.447 | 0.841 | 0.875 | Uncertain | 1 | 6-33451776-C-T | 3 | 1.86e-6 | -4.152 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -1.63 | Neutral | 0.017 | Benign | 0.028 | Benign | 2.83 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.1886 | 0.4655 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.3931C>A | L1311M 2D AIThe SynGAP1 missense variant L1311M is reported in gnomAD (ID 6‑33451805‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates “Likely Benign.” In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign classification for L1311M, and this conclusion is not contradicted by any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.968153 | Binding | 0.393 | 0.907 | 0.750 | 6-33451805-C-A | 3 | 1.86e-6 | -4.817 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.060 | Likely Benign | 0.18 | Neutral | 0.936 | Possibly Damaging | 0.632 | Possibly Damaging | 2.73 | Benign | 0.23 | Tolerated | 3.77 | 5 | 0.1068 | 0.4279 | 2 | 4 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||
| c.1169G>C | G390A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G390A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and Rosetta. The high‑accuracy assessment shows AlphaMissense‑Optimized as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as pathogenic. FoldX alone is inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -6.768 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 1.96 | Ambiguous | 0.4 | 2.03 | Destabilizing | 2.00 | Destabilizing | 0.02 | Likely Benign | 0.402 | Likely Benign | -0.55 | Neutral | 0.143 | Benign | 0.028 | Benign | 1.33 | Pathogenic | 0.05 | Affected | 0.4130 | 0.5053 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1195G>T | A399S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A399S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect, while FoldX, Rosetta, and Foldetta are inconclusive. Grouping by agreement, all available predictors fall into the benign category; none predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, whereas Foldetta’s stability analysis remains uncertain. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | -4.256 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.1 | 1.13 | Ambiguous | 0.89 | Ambiguous | -0.33 | Likely Benign | 0.161 | Likely Benign | 0.81 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.65 | Benign | 0.86 | Tolerated | 0.2494 | 0.4897 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1994A>T | Y665F 2D 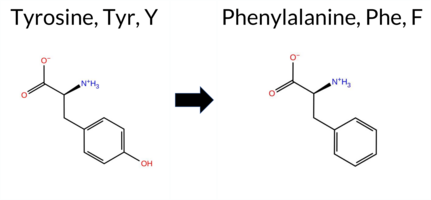 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while Rosetta and Foldetta are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar claim; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | -8.460 | Likely Pathogenic | 0.100 | Likely Benign | Likely Benign | 0.07 | Likely Benign | 0.1 | 1.03 | Ambiguous | 0.55 | Ambiguous | 0.38 | Likely Benign | 0.106 | Likely Benign | -1.23 | Neutral | 0.159 | Benign | 0.036 | Benign | 3.45 | Benign | 0.47 | Tolerated | 0.2277 | 0.3076 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||
| c.2230C>G | Q744E 2D AIThe SynGAP1 missense variant Q744E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” while Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -4.053 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.57 | Neutral | 0.065 | Benign | 0.038 | Benign | 2.77 | Benign | 0.08 | Tolerated | 0.1453 | 0.1892 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2231A>T | Q744L 2D AIThe SynGAP1 missense variant Q744L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the preponderance of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -3.724 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -1.96 | Neutral | 0.425 | Benign | 0.158 | Benign | 2.70 | Benign | 0.02 | Affected | 0.0813 | 0.4731 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.227C>G | S76C 2D AIThe SynGAP1 missense variant S76C is listed in ClinVar with an “Uncertain” status (ClinVar ID 1951273.0) and is present in the gnomAD database (gnomAD ID 6‑33425835‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as benign; Foldetta results are not available for this variant. Overall, the majority of computational evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | Uncertain | 1 | 6-33425835-C-G | 2 | 1.24e-6 | -5.408 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -1.78 | Neutral | 0.992 | Probably Damaging | 0.869 | Possibly Damaging | 3.71 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0807 | 0.4787 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||
| c.2566A>C | N856H 2D 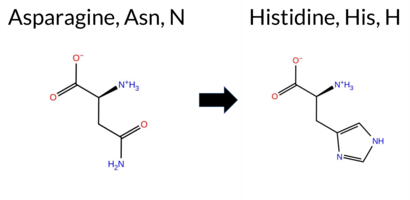 AIThe SynGAP1 missense variant N856H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions, including the high‑accuracy tools, suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.477615 | Uncertain | 0.263 | 0.827 | 0.500 | -2.596 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -1.26 | Neutral | 0.990 | Probably Damaging | 0.923 | Probably Damaging | 4.10 | Benign | 0.09 | Tolerated | 0.1674 | 0.7495 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2876A>T | H959L 2D AIThe SynGAP1 missense variant H959L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H959L is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | -8.515 | Likely Pathogenic | 0.100 | Likely Benign | Likely Benign | 0.236 | Likely Benign | -1.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.14 | Benign | 0.09 | Tolerated | 0.1698 | 0.5538 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2898C>A | H966Q 2D 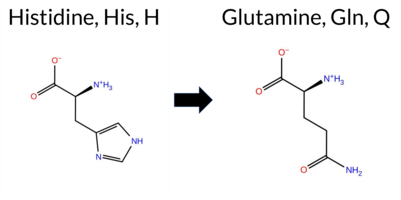 AIThe SynGAP1 missense variant H966Q is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that H966Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -5.662 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.66 | Neutral | 0.748 | Possibly Damaging | 0.232 | Benign | 4.06 | Benign | 0.45 | Tolerated | 0.2058 | 0.3914 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2898C>G | H966Q 2D AIThe SynGAP1 missense variant H966Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -5.662 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.66 | Neutral | 0.748 | Possibly Damaging | 0.232 | Benign | 4.06 | Benign | 0.45 | Tolerated | 0.2058 | 0.3914 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3169A>T | S1057C 2D AIThe SynGAP1 missense variant S1057C is reported in gnomAD (ID 6‑33443721‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic impact, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | 6-33443721-A-T | -7.529 | In-Between | 0.100 | Likely Benign | Likely Benign | 0.258 | Likely Benign | -0.64 | Neutral | 0.977 | Probably Damaging | 0.683 | Possibly Damaging | 5.23 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1856 | 0.6106 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3215A>G | K1072R 2D AIThe SynGAP1 missense variant K1072R is reported in gnomAD (ID 6‑33443767‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. When predictions are grouped by consensus, the benign group contains seven tools, whereas the pathogenic group contains two. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized reports a benign outcome, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No Foldetta stability data are available, so folding‑stability evidence is unavailable. Overall, the majority of evidence points to a benign impact, and this is not in conflict with ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | 6-33443767-A-G | 1 | 6.23e-7 | -2.458 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.15 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 4.16 | Benign | 0.88 | Tolerated | 3.77 | 5 | 0.4655 | 0.2061 | 2 | 3 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||
| c.3298G>A | G1100S 2D AIThe SynGAP1 missense variant G1100S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for G1100S, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.898 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.65 | Neutral | 0.943 | Possibly Damaging | 0.595 | Possibly Damaging | 2.10 | Pathogenic | 0.28 | Tolerated | 0.2501 | 0.5699 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3424T>A | S1142T 2D AIThe SynGAP1 missense variant S1142T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.719935 | Binding | 0.276 | 0.844 | 1.000 | -3.712 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -1.63 | Neutral | 0.611 | Possibly Damaging | 0.324 | Benign | 2.69 | Benign | 0.00 | Affected | 0.1548 | 0.6427 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3671T>A | L1224Q 2D AIThe SynGAP1 missense variant L1224Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports likely benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for L1224Q, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -6.254 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.87 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.40 | Pathogenic | 0.13 | Tolerated | 0.1018 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.37A>C | I13L 2D AIThe SynGAP1 missense variant I13L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.482657 | Uncertain | 0.318 | 0.916 | 0.375 | -2.144 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.095 | Likely Benign | 0.07 | Neutral | 0.002 | Benign | 0.001 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1055 | 0.4923 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3854C>A | P1285Q 2D AIThe SynGAP1 missense variant P1285Q is reported in gnomAD (variant ID 6‑33447902‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is provided). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.821643 | Binding | 0.557 | 0.759 | 0.750 | 6-33447902-C-A | -4.073 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -1.67 | Neutral | 0.977 | Probably Damaging | 0.632 | Possibly Damaging | 4.22 | Benign | 0.13 | Tolerated | 4.32 | 2 | 0.1476 | 0.3025 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||
| c.3911C>T | P1304L 2D AIThe SynGAP1 missense variant P1304L is reported in gnomAD (ID 6‑33451785‑C‑T) and has no ClinVar entry. All evaluated in silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus (SGM‑Consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome, while AlphaMissense‑Optimized independently scores it benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Based on the unanimous benign predictions and the absence of any ClinVar pathogenic classification, the variant is most likely benign and does not contradict existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.886417 | Binding | 0.475 | 0.866 | 0.875 | 6-33451785-C-T | 1 | 6.20e-7 | -4.080 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -1.33 | Neutral | 0.126 | Benign | 0.066 | Benign | 2.83 | Benign | 0.06 | Tolerated | 0.2200 | 0.5478 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||
| c.3932T>A | L1311Q 2D AIThe SynGAP1 missense variant L1311Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.968153 | Binding | 0.393 | 0.907 | 0.750 | -4.009 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.03 | Neutral | 0.579 | Possibly Damaging | 0.413 | Benign | 2.74 | Benign | 0.12 | Tolerated | 0.1476 | 0.1677 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.1033C>A | L345M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L345M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a benign outcome; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is uncertain. Overall, the majority of evidence points to a benign impact for the L345M variant, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -3.918 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.75 | Ambiguous | 0.1 | 0.80 | Ambiguous | 0.78 | Ambiguous | 0.54 | Ambiguous | 0.101 | Likely Benign | -0.97 | Neutral | 0.802 | Possibly Damaging | 0.122 | Benign | 1.73 | Pathogenic | 0.07 | Tolerated | 0.0677 | 0.3246 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.124C>A | P42T 2D AIThe SynGAP1 missense variant P42T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P42T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.626 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.97 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.29 | Benign | 0.00 | Affected | 0.1834 | 0.4896 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.1328G>C | G443A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach a consensus all indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. The protein‑folding stability predictor Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result and is therefore treated as unavailable evidence. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -0.332 | Likely Benign | 0.101 | Likely Benign | Likely Benign | -0.59 | Ambiguous | 0.1 | -1.61 | Ambiguous | -1.10 | Ambiguous | -0.52 | Ambiguous | 0.033 | Likely Benign | -1.04 | Neutral | 0.022 | Benign | 0.011 | Benign | 3.41 | Benign | 0.54 | Tolerated | 0.3539 | 0.3192 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1832T>C | M611T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611T is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33440884‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. Four tools (FoldX, Rosetta, Foldetta, premPS) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | Uncertain | 1 | 6-33440884-T-C | 1 | 6.19e-7 | -5.696 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 1.98 | Ambiguous | 0.2 | 0.94 | Ambiguous | 1.46 | Ambiguous | 0.87 | Ambiguous | 0.240 | Likely Benign | -2.40 | Neutral | 0.034 | Benign | 0.038 | Benign | -1.19 | Pathogenic | 0.29 | Tolerated | 3.37 | 35 | 0.1635 | 0.1415 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||
| c.1904A>G | N635S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N635S is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6-33440956-A-G). Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and ESM1b. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta is also inconclusive. Overall, the majority of available predictions lean toward a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | Conflicting | 4 | 6-33440956-A-G | 10 | 6.20e-6 | -9.002 | Likely Pathogenic | 0.101 | Likely Benign | Likely Benign | 0.80 | Ambiguous | 0.1 | 0.67 | Ambiguous | 0.74 | Ambiguous | 0.95 | Ambiguous | 0.104 | Likely Benign | -4.45 | Deleterious | 0.261 | Benign | 0.044 | Benign | 3.06 | Benign | 0.05 | Affected | 3.37 | 34 | 0.2816 | 0.4279 | 1 | 1 | 2.7 | -27.03 | 196.0 | 30.9 | 0.1 | 0.0 | -0.3 | 0.2 | X | Uncertain | In the WT simulations, the carboxamide side chain of Asn635, located on the outer surface of an α helix (res. Glu617-Asn635), forms hydrogen bonds with Gln631 on the same α helix and with the hydroxyl side chain of Ser590 on an opposing α helix (res. Glu582-Met603).In the variant simulations, the side chain of Ser635 is shorter than asparagine and thus prefers to hydrogen bond with the carbonyl group of Gln631 on the same helix and, to a lesser extent, with Ser590 compared to Asn635 in the WT. Ser635 forms hydrogen bonds with the backbone atoms of the same helix, which may destabilize the helix, although this is not clearly evident in the simulations. The weakening of the hydrogen bond between Ser635 and Ser590 in the variant may also weaken the tertiary structure assembly between the helices.Additionally, Asn635 is at the GTPase interface. However, the implication of the residue swap on the complex formation with the GTPase cannot be investigated using solvent-only simulations. | ||||||||||||||
| c.2125C>G | L709V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709V is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool that produced a definitive call classifies the variant as benign: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. The only inconclusive results are FoldX (uncertain) and Foldetta (uncertain), which are treated as unavailable evidence. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also benign; Foldetta remains uncertain. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -4.406 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.75 | Ambiguous | 0.0 | 0.42 | Likely Benign | 0.59 | Ambiguous | 0.03 | Likely Benign | 0.063 | Likely Benign | -0.36 | Neutral | 0.062 | Benign | 0.016 | Benign | 3.42 | Benign | 0.45 | Tolerated | 0.1162 | 0.2460 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2224C>G | R742G 2D AIThe SynGAP1 missense variant R742G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | -4.065 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.25 | Neutral | 0.524 | Possibly Damaging | 0.259 | Benign | 2.70 | Benign | 0.02 | Affected | 0.3974 | 0.2805 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2225G>T | R742L 2D  AIThe SynGAP1 missense variant R742L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | -3.778 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -0.77 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.71 | Benign | 0.16 | Tolerated | 0.2342 | 0.3831 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2251C>A | P751T 2D AIThe SynGAP1 missense variant P751T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -5.111 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -1.65 | Neutral | 0.679 | Possibly Damaging | 0.348 | Benign | 2.71 | Benign | 1.00 | Tolerated | 0.1613 | 0.5704 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2254T>A | S752T 2D AIThe SynGAP1 missense variant S752T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -4.040 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -1.33 | Neutral | 0.248 | Benign | 0.137 | Benign | 1.55 | Pathogenic | 0.07 | Tolerated | 0.1633 | 0.6342 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.230G>A | S77N 2D  AIThe SynGAP1 missense variant S77N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -4.597 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.78 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.09 | Benign | 0.00 | Affected | 0.1088 | 0.3774 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.2381C>A | P794Q 2D AIThe SynGAP1 missense variant P794Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for P794Q, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | -4.641 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.31 | Neutral | 0.918 | Possibly Damaging | 0.601 | Possibly Damaging | 4.24 | Benign | 0.02 | Affected | 0.1581 | 0.4971 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||
| c.2597T>C | V866A 2D  AIThe SynGAP1 missense variant V866A is reported in gnomAD (ID 6‑33443149‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.638070 | Binding | 0.266 | 0.788 | 0.250 | 6-33443149-T-C | 1 | 6.20e-7 | -1.805 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -1.27 | Neutral | 0.889 | Possibly Damaging | 0.682 | Possibly Damaging | 2.87 | Benign | 0.37 | Tolerated | 3.82 | 4 | 0.3441 | 0.2512 | 0 | 0 | -2.4 | -28.05 | ||||||||||||||||||||||||||||||||||
| c.2643G>C | L881F 2D AIThe SynGAP1 missense variant L881F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for L881F, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.629350 | Binding | 0.299 | 0.874 | 0.250 | -5.759 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.87 | Neutral | 0.818 | Possibly Damaging | 0.360 | Benign | 2.48 | Pathogenic | 0.02 | Affected | 0.0711 | 0.3415 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2643G>T | L881F 2D AIThe SynGAP1 missense variant L881F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for L881F, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.629350 | Binding | 0.299 | 0.874 | 0.250 | -5.759 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.87 | Neutral | 0.818 | Possibly Damaging | 0.360 | Benign | 2.48 | Pathogenic | 0.02 | Affected | 0.0711 | 0.3415 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2878C>A | H960N 2D AIThe SynGAP1 missense variant H960N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987911 | Disordered | 0.983385 | Binding | 0.380 | 0.901 | 0.750 | -8.822 | Likely Pathogenic | 0.101 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -0.57 | Neutral | 0.494 | Possibly Damaging | 0.129 | Benign | 4.19 | Benign | 0.40 | Tolerated | 0.2137 | 0.3695 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2895C>A | H965Q 2D AIThe SynGAP1 missense variant H965Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.447 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -0.71 | Neutral | 0.138 | Benign | 0.105 | Benign | 4.09 | Benign | 0.28 | Tolerated | 0.2257 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2895C>G | H965Q 2D AIThe SynGAP1 missense variant H965Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classify the variant as Likely Benign. Individual algorithms—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—all predict a benign outcome. No tool in the dataset returned a pathogenic prediction. High‑accuracy methods: AlphaMissense‑Optimized is benign; SGM‑Consensus is Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.447 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.71 | Neutral | 0.138 | Benign | 0.105 | Benign | 4.09 | Benign | 0.28 | Tolerated | 0.2257 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2993C>T | A998V 2D AIThe SynGAP1 missense variant A998V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -4.795 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -1.09 | Neutral | 0.245 | Benign | 0.138 | Benign | 4.09 | Benign | 0.00 | Affected | 0.1281 | 0.6147 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.302A>T | H101L 2D AIThe SynGAP1 missense variant H101L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for H101L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.688356 | Binding | 0.370 | 0.884 | 0.625 | -1.376 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.33 | Neutral | 0.824 | Possibly Damaging | 0.840 | Possibly Damaging | 4.19 | Benign | 0.00 | Affected | 0.0924 | 0.4960 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3052A>C | T1018P 2D AIThe SynGAP1 missense variant T1018P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.959985 | Binding | 0.348 | 0.801 | 0.500 | -2.046 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.218 | Likely Benign | -1.39 | Neutral | 0.586 | Possibly Damaging | 0.302 | Benign | 2.24 | Pathogenic | 0.02 | Affected | 0.2155 | 0.3960 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3296A>T | Y1099F 2D AIThe SynGAP1 missense variant Y1099F is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -3.651 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.85 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.75 | Benign | 0.27 | Tolerated | 0.2453 | 0.2780 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.370G>A | A124T 2D AIThe SynGAP1 missense variant A124T is reported in gnomAD (6-33432235‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Taken together, the preponderance of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.699139 | Binding | 0.340 | 0.883 | 0.750 | 6-33432235-G-A | 1 | 6.20e-7 | -3.551 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.26 | Neutral | 0.734 | Possibly Damaging | 0.187 | Benign | 4.17 | Benign | 0.78 | Tolerated | 3.61 | 5 | 0.1828 | 0.7680 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.370G>T | A124S 2D AIThe SynGAP1 missense variant A124 S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.699139 | Binding | 0.340 | 0.883 | 0.750 | -1.933 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.053 | Likely Benign | 0.35 | Neutral | 0.849 | Possibly Damaging | 0.303 | Benign | 4.22 | Benign | 0.65 | Tolerated | 0.3018 | 0.6192 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.991T>G | S331A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S331A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority vote (3 benign vs. 1 pathogenic). High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote) is benign; and Foldetta, combining FoldX‑MD and Rosetta outputs, is benign. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.433034 | Structured | 0.346458 | Uncertain | 0.658 | 0.475 | 0.250 | -2.199 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.1 | -0.08 | Likely Benign | 0.07 | Likely Benign | -0.15 | Likely Benign | 0.071 | Likely Benign | -0.44 | Neutral | 0.139 | Benign | 0.060 | Benign | 1.89 | Pathogenic | 0.67 | Tolerated | 0.5314 | 0.3008 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||
| c.1609G>C | A537P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A537P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FATHMM and Rosetta; FoldX and Foldetta are inconclusive. The high‑accuracy consensus (SGM‑Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN indicates a likely benign outcome, while AlphaMissense‑Optimized also predicts benign. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the majority of evidence points to a benign impact for A537P, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.037313 | Uncertain | 0.928 | 0.362 | 0.000 | 0.404 | Likely Benign | 0.102 | Likely Benign | Likely Benign | -0.61 | Ambiguous | 0.6 | 2.43 | Destabilizing | 0.91 | Ambiguous | -0.18 | Likely Benign | 0.218 | Likely Benign | 0.67 | Neutral | 0.020 | Benign | 0.022 | Benign | -1.29 | Pathogenic | 0.35 | Tolerated | 0.2152 | 0.3807 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.2080G>A | A694T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A694T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Taken together, the overwhelming majority of evidence indicates a benign effect for A694T, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -3.565 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.1 | 0.11 | Likely Benign | 0.33 | Likely Benign | -0.26 | Likely Benign | 0.095 | Likely Benign | -0.71 | Neutral | 0.787 | Possibly Damaging | 0.098 | Benign | 3.46 | Benign | 0.17 | Tolerated | 0.1618 | 0.5440 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.2203A>T | S735C 2D AIThe SynGAP1 missense variant S735C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S735C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -7.291 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -2.22 | Neutral | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.60 | Benign | 0.05 | Affected | 0.1136 | 0.5464 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.236A>C | N79T 2D AIThe SynGAP1 missense variant N79T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -3.752 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.022 | Likely Benign | -0.61 | Neutral | 0.371 | Benign | 0.018 | Benign | 4.21 | Benign | 0.00 | Affected | 0.1133 | 0.4890 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.2552C>A | P851H 2D AIThe SynGAP1 missense variant P851H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.648219 | Disordered | 0.526893 | Binding | 0.347 | 0.819 | 0.625 | -4.730 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.184 | Likely Benign | 0.19 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 4.18 | Benign | 0.15 | Tolerated | 0.1577 | 0.5378 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2647C>A | L883I 2D AIThe SynGAP1 missense variant L883I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.641952 | Binding | 0.334 | 0.886 | 0.250 | -5.046 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -0.26 | Neutral | 0.802 | Possibly Damaging | 0.355 | Benign | 2.66 | Benign | 0.35 | Tolerated | 0.0970 | 0.4093 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2842G>T | G948C 2D AIThe SynGAP1 missense variant G948C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar assertion exists for this variant. Thus, based on the available computational predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.862121 | Binding | 0.365 | 0.919 | 0.750 | -10.254 | Likely Pathogenic | 0.102 | Likely Benign | Likely Benign | 0.247 | Likely Benign | -0.77 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 4.50 | Benign | 0.04 | Affected | 0.1422 | 0.4439 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2843G>T | G948V 2D AIThe SynGAP1 missense variant G948V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that G948V is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.862121 | Binding | 0.365 | 0.919 | 0.750 | -6.541 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.256 | Likely Benign | -0.48 | Neutral | 0.901 | Possibly Damaging | 0.435 | Benign | 4.52 | Benign | 0.06 | Tolerated | 0.1464 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2846G>T | G949V 2D AIThe SynGAP1 missense variant G949V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -7.364 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.328 | Likely Benign | -0.55 | Neutral | 0.992 | Probably Damaging | 0.834 | Possibly Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1493 | 0.3165 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2892C>A | H964Q 2D AIThe SynGAP1 missense variant H964Q is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The only tool with an uncertain call is ESM1b, and no pathogenic predictions are reported. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.279 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.18 | Benign | 0.07 | Tolerated | 0.1969 | 0.3593 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2892C>G | H964Q 2D AIThe SynGAP1 missense variant H964Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only uncertain result comes from ESM1b. The high‑accuracy consensus methods also support a benign classification: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the evidence overwhelmingly indicates that H964Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.279 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.18 | Benign | 0.07 | Tolerated | 0.1969 | 0.3593 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3152G>T | G1051V 2D AIThe SynGAP1 missense variant G1051V is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443704‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction (2 benign vs. 1 pathogenic, 1 uncertain). Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for G1051V, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | 6-33443704-G-T | 1 | 6.20e-7 | -7.098 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.460 | Likely Benign | -0.62 | Neutral | 0.245 | Benign | 0.096 | Benign | -0.74 | Pathogenic | 0.17 | Tolerated | 3.77 | 5 | 0.1274 | 0.3680 | -3 | -1 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||
| c.3347G>T | G1116V 2D AIThe SynGAP1 missense variant G1116V is reported in gnomAD (variant ID 6‑33443899‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443899-G-T | -6.426 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.393 | Likely Benign | -0.79 | Neutral | 0.626 | Possibly Damaging | 0.375 | Benign | 4.06 | Benign | 0.06 | Tolerated | 4.32 | 2 | 0.1268 | 0.3494 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.3353G>A | S1118N 2D AIThe SynGAP1 missense variant S1118N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | -6.551 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.223 | Likely Benign | -0.53 | Neutral | 0.901 | Possibly Damaging | 0.433 | Benign | 5.26 | Benign | 0.06 | Tolerated | 0.1924 | 0.4215 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3925G>C | V1309L 2D AIThe SynGAP1 missense variant V1309L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.948596 | Binding | 0.402 | 0.907 | 0.750 | -2.543 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.108 | Likely Benign | 1.41 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.99 | Benign | 1.00 | Tolerated | 0.0944 | 0.4212 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3925G>T | V1309L 2D AIThe SynGAP1 missense variant V1309L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the computational evidence strongly supports a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.948596 | Binding | 0.402 | 0.907 | 0.750 | -2.543 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.108 | Likely Benign | 1.41 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.99 | Benign | 1.00 | Tolerated | 0.0944 | 0.4212 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.4018A>C | T1340P 2D AIThe SynGAP1 missense variant T1340P is catalogued in gnomAD (ID 6‑33451892‑A‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes, while only SIFT predicts a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that T1340P is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.977899 | Binding | 0.444 | 0.697 | 0.750 | 6-33451892-A-C | -2.681 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.190 | Likely Benign | -1.81 | Neutral | 0.334 | Benign | 0.099 | Benign | 4.08 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2268 | 0.4983 | -1 | 0 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||||||||
| c.676T>A | S226T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S226T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while FoldX is uncertain and therefore treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.129801 | Structured | 0.334959 | Uncertain | 0.800 | 0.324 | 0.250 | -5.066 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.53 | Ambiguous | 0.2 | 0.12 | Likely Benign | 0.33 | Likely Benign | -0.10 | Likely Benign | 0.326 | Likely Benign | -1.47 | Neutral | 0.390 | Benign | 0.079 | Benign | 5.75 | Benign | 0.05 | Affected | 0.1991 | 0.6681 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1069C>A | H357N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H357N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Taken together, the overwhelming majority of evidence points to a benign impact. There is no ClinVar annotation to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -7.617 | In-Between | 0.103 | Likely Benign | Likely Benign | 0.14 | Likely Benign | 0.2 | 0.31 | Likely Benign | 0.23 | Likely Benign | 0.31 | Likely Benign | 0.126 | Likely Benign | -1.47 | Neutral | 0.495 | Possibly Damaging | 0.169 | Benign | 4.25 | Benign | 0.15 | Tolerated | 0.2052 | 0.3266 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.1142G>C | G381A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G381A is reported in gnomAD (variant ID 6-33438047‑G‑C) but has no ClinVar entry. Functional prediction tools fall into two groups: benign predictions come from premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar; pathogenic predictions come from REVEL, FoldX, Rosetta, polyPhen‑2 HumDiv, FATHMM, and the SGM‑Consensus score. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, while Foldetta (combining FoldX‑MD and Rosetta stability outputs) indicates a pathogenic effect. No prediction or stability result is missing or inconclusive. Overall, the majority of tools suggest a benign effect, and the high‑accuracy consensus leans toward benign, though Foldetta’s pathogenic signal introduces uncertainty. The variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | 6-33438047-G-C | 1 | 6.23e-7 | -6.266 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 3.97 | Destabilizing | 0.7 | 2.05 | Destabilizing | 3.01 | Destabilizing | 0.06 | Likely Benign | 0.507 | Likely Pathogenic | -0.63 | Neutral | 0.718 | Possibly Damaging | 0.332 | Benign | 1.33 | Pathogenic | 0.52 | Tolerated | 4.32 | 9 | 0.3809 | 0.4770 | 0 | 1 | 2.2 | 14.03 | ||||||||||||||||||||||||
| c.1187G>C | G396A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -3.655 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 1.74 | Ambiguous | 0.7 | 1.90 | Ambiguous | 1.82 | Ambiguous | 0.41 | Likely Benign | 0.274 | Likely Benign | -1.28 | Neutral | 0.062 | Benign | 0.024 | Benign | 3.93 | Benign | 0.84 | Tolerated | 0.3846 | 0.5255 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.2156A>C | N719T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign) all predict a neutral impact. In contrast, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar predict a pathogenic effect, while Rosetta remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -6.251 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.0 | -0.59 | Ambiguous | -0.10 | Likely Benign | 0.44 | Likely Benign | 0.078 | Likely Benign | -2.60 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.76 | Benign | 0.38 | Tolerated | 0.0851 | 0.4620 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.2186A>C | N729T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729T is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID 6‑33441651‑A‑C). Consensus among the majority of in‑silico predictors is benign: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, yields an uncertain result. Overall, the evidence strongly supports a benign effect, and this conclusion does not contradict ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | 6-33441651-A-C | 1 | 6.20e-7 | -1.952 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.3 | 1.73 | Ambiguous | 1.13 | Ambiguous | -0.34 | Likely Benign | 0.052 | Likely Benign | -0.52 | Neutral | 0.123 | Benign | 0.042 | Benign | 3.33 | Benign | 1.00 | Tolerated | 3.59 | 7 | 0.1201 | 0.4805 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||
| c.25C>A | H9N 2D AIThe SynGAP1 missense variant H9N is reported in gnomAD (ID 6‑33420289‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | 6-33420289-C-A | -3.789 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -0.36 | Neutral | 0.024 | Benign | 0.003 | Benign | 4.23 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2327 | 0.3823 | 1 | 2 | -0.3 | -23.04 | ||||||||||||||||||||||||||||||||||||
| c.2744G>C | G915A 2D AIThe SynGAP1 missense variant G915A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for G915A, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.808641 | Binding | 0.302 | 0.880 | 0.375 | -4.007 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -1.22 | Neutral | 0.900 | Possibly Damaging | 0.622 | Possibly Damaging | 2.87 | Benign | 0.05 | Affected | 0.3454 | 0.4930 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2987C>A | P996H 2D AIThe SynGAP1 missense variant P996H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | -5.554 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -1.19 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1585 | 0.5160 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3037T>C | S1013P 2D AIThe SynGAP1 missense variant S1013P is reported in gnomAD (ID 6‑33443589‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, while the consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.899570 | Binding | 0.308 | 0.846 | 0.625 | 6-33443589-T-C | 2 | 1.24e-6 | -2.563 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -1.27 | Neutral | 0.453 | Possibly Damaging | 0.150 | Benign | 2.66 | Benign | 0.15 | Tolerated | 3.77 | 5 | 0.2338 | 0.5430 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.3179G>T | G1060V 2D AIThe SynGAP1 missense variant G1060V is listed in ClinVar as benign (ClinVar ID 1345112.0) and is observed in gnomAD (6‑33443731‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely benign, and AlphaMissense‑Optimized also reports a benign outcome. No Foldetta stability assessment is available for this variant. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar designation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | Benign | 1 | 6-33443731-G-T | 1 | 6.22e-7 | -6.966 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.369 | Likely Benign | -0.73 | Neutral | 0.986 | Probably Damaging | 0.728 | Possibly Damaging | 2.63 | Benign | 0.33 | Tolerated | 4.32 | 2 | 0.1453 | 0.3494 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.3185G>T | G1062V 2D AIThe SynGAP1 missense variant G1062V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -6.598 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.377 | Likely Benign | -0.78 | Neutral | 0.259 | Benign | 0.066 | Benign | 4.12 | Benign | 0.01 | Affected | 0.1441 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.328G>A | V110I 2D AIThe SynGAP1 missense variant V110I is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.409 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.10 | Neutral | 0.012 | Benign | 0.006 | Benign | 4.26 | Benign | 0.52 | Tolerated | 0.0875 | 0.4485 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3674C>G | S1225C 2D AIThe SynGAP1 missense variant S1225C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign. Foldetta results are not available for this variant. Overall, the preponderance of evidence indicates that S1225C is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.513880 | Disordered | 0.441915 | Uncertain | 0.891 | 0.544 | 0.500 | -6.494 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.306 | Likely Benign | -0.27 | Neutral | 0.975 | Probably Damaging | 0.870 | Possibly Damaging | 5.35 | Benign | 0.15 | Tolerated | 0.0669 | 0.4842 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||
| c.3871C>G | L1291V 2D AIThe SynGAP1 missense variant L1291V is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity, so the pathogenic prediction group is empty. High‑accuracy assessments corroborate this benign consensus: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this prediction aligns with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.863458 | Binding | 0.579 | 0.797 | 0.625 | -4.583 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.028 | Likely Benign | 0.28 | Neutral | 0.149 | Benign | 0.064 | Benign | 2.61 | Benign | 0.07 | Tolerated | 0.1435 | 0.2946 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.674C>T | S225L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S225L is reported in gnomAD (6‑33435525‑C‑T) but has no ClinVar entry. In silico predictors that agree on a benign effect include REVEL, FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only PROVEAN predicts a pathogenic outcome. Predictions that are inconclusive are Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign effect for S225L, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.150080 | Structured | 0.344191 | Uncertain | 0.817 | 0.319 | 0.250 | 6-33435525-C-T | 1 | 6.20e-7 | -6.644 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.29 | Likely Benign | 0.4 | 1.18 | Ambiguous | 0.74 | Ambiguous | 0.18 | Likely Benign | 0.320 | Likely Benign | -3.76 | Deleterious | 0.000 | Benign | 0.001 | Benign | 5.70 | Benign | 0.28 | Tolerated | 3.41 | 13 | 0.1039 | 0.6554 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||
| c.79C>A | P27T 2D AIThe SynGAP1 missense variant P27T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence, including high‑accuracy tools, points to a benign effect for P27T, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.437871 | Uncertain | 0.430 | 0.881 | 0.375 | -3.612 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -2.07 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 3.85 | Benign | 0.00 | Affected | 0.2430 | 0.5923 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.112C>T | P38S 2D AIThe SynGAP1 missense variant P38S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -2.727 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -2.13 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.3837 | 0.6111 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.1160G>C | G387A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G387A is reported in gnomAD (variant ID 6‑33438065‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts it as pathogenic. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive (treated as unavailable). No other tools provide decisive evidence. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.422910 | Uncertain | 0.293 | 0.861 | 0.750 | 6-33438065-G-C | 3 | 1.87e-6 | -6.313 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 1.95 | Ambiguous | 0.1 | 1.68 | Ambiguous | 1.82 | Ambiguous | -0.02 | Likely Benign | 0.300 | Likely Benign | -0.29 | Neutral | 0.007 | Benign | 0.010 | Benign | 1.33 | Pathogenic | 0.06 | Tolerated | 4.32 | 3 | 0.4149 | 0.4576 | 0 | 1 | 2.2 | 14.03 | ||||||||||||||||||||||||
| c.1288A>C | M430L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, FoldX, Rosetta, PolyPhen‑2 (HumDiv and HumVar), SIFT, and REVEL; no tool predicts pathogenicity. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. With all available evidence pointing to a benign effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -5.873 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.33 | Likely Benign | 0.1 | -0.10 | Likely Benign | 0.12 | Likely Benign | 0.72 | Ambiguous | 0.107 | Likely Benign | -1.07 | Neutral | 0.028 | Benign | 0.004 | Benign | 3.69 | Benign | 0.66 | Tolerated | 0.1767 | 0.5067 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1288A>T | M430L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, FoldX, Rosetta, PolyPhen‑2 (HumDiv and HumVar), SIFT, and REVEL; no tool predicts pathogenicity. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. With all available evidence pointing to a benign effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -5.873 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.33 | Likely Benign | 0.1 | -0.10 | Likely Benign | 0.12 | Likely Benign | 0.72 | Ambiguous | 0.107 | Likely Benign | -1.07 | Neutral | 0.028 | Benign | 0.004 | Benign | 3.69 | Benign | 0.66 | Tolerated | 0.1767 | 0.5067 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1600T>A | S534T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S534T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate benign or likely benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Thus, the overall evidence strongly supports a benign classification, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.167087 | Structured | 0.032173 | Uncertain | 0.860 | 0.362 | 0.000 | -4.925 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.28 | Likely Benign | 0.19 | Likely Benign | 0.200 | Likely Benign | -2.42 | Neutral | 0.676 | Possibly Damaging | 0.933 | Probably Damaging | 3.32 | Benign | 0.07 | Tolerated | 0.1238 | 0.5064 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2513A>G | N838S 2D AIThe SynGAP1 missense variant at residue N838S is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Only the two polyPhen‑2 classifiers (HumDiv and HumVar) predict pathogenicity. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign classification, and this is not in conflict with ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.613320 | Binding | 0.276 | 0.861 | 0.250 | -3.748 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -1.24 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.83 | Benign | 0.50 | Tolerated | 0.3552 | 0.5485 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.2542G>A | G848S 2D AIThe SynGAP1 missense variant G848S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that G848S is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.563942 | Binding | 0.287 | 0.816 | 0.500 | -4.077 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.134 | Likely Benign | 0.07 | Neutral | 0.856 | Possibly Damaging | 0.476 | Possibly Damaging | 2.62 | Benign | 0.11 | Tolerated | 0.2809 | 0.4734 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2705C>G | A902G 2D AIThe SynGAP1 missense variant A902G is not listed in ClinVar and has no entry in gnomAD, indicating no reported clinical classification or population frequency data. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT predicts a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the consensus of available predictions points to a benign impact, with no conflict with ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.517703 | Binding | 0.319 | 0.919 | 0.375 | -4.046 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.020 | Likely Benign | -0.65 | Neutral | 0.004 | Benign | 0.008 | Benign | 2.69 | Benign | 0.03 | Affected | 0.2177 | 0.4461 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2875C>A | H959N 2D AIThe SynGAP1 missense variant H959N is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only ESM1b predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple independent predictors and high‑accuracy tools indicates that H959N is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | -8.811 | Likely Pathogenic | 0.104 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -0.10 | Neutral | 0.144 | Benign | 0.058 | Benign | 4.15 | Benign | 0.21 | Tolerated | 0.2298 | 0.3838 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.294T>A | H98Q 2D AIThe SynGAP1 missense variant H98Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -2.749 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.47 | Neutral | 0.002 | Benign | 0.000 | Benign | 4.26 | Benign | 0.00 | Affected | 0.1831 | 0.4347 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.294T>G | H98Q 2D AIThe SynGAP1 missense variant H98Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -2.749 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.47 | Neutral | 0.002 | Benign | 0.000 | Benign | 4.26 | Benign | 0.00 | Affected | 0.1831 | 0.4347 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2951A>G | K984R 2D AIThe SynGAP1 missense variant K984R is reported in gnomAD (ID 6‑33443503‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of tools indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.951648 | Binding | 0.288 | 0.895 | 0.750 | 6-33443503-A-G | 2 | 1.24e-6 | -2.044 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.41 | Neutral | 0.012 | Benign | 0.012 | Benign | 2.67 | Benign | 0.00 | Affected | 4.32 | 1 | 0.5098 | 0.1558 | Weaken | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||
| c.2974G>T | V992F 2D  AIThe SynGAP1 missense variant V992F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and there is no conflict with ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -3.131 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.25 | Neutral | 0.680 | Possibly Damaging | 0.356 | Benign | 4.17 | Benign | 0.04 | Affected | 0.0832 | 0.4083 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.3027G>C | E1009D 2D  AIThe SynGAP1 missense variant E1009D is reported in gnomAD (ID 6‑33443579‑G‑C) but has no ClinVar entry. All in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized indicates benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign effect, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | 6-33443579-G-C | 1 | 6.20e-7 | -2.958 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.37 | Neutral | 0.011 | Benign | 0.017 | Benign | 2.50 | Benign | 0.30 | Tolerated | 3.77 | 5 | 0.2077 | 0.4817 | 2 | 3 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.3027G>T | E1009D 2D AIThe SynGAP1 missense variant E1009D is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -2.958 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.37 | Neutral | 0.011 | Benign | 0.017 | Benign | 2.50 | Benign | 0.30 | Tolerated | 3.77 | 5 | 0.2077 | 0.4817 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.3035C>G | P1012R 2D  AIThe SynGAP1 missense variant P1012R is not reported in ClinVar and is absent from gnomAD, indicating no documented population frequency. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.894674 | Binding | 0.319 | 0.866 | 0.625 | -4.413 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.034 | Likely Benign | 1.24 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.89 | Benign | 0.88 | Tolerated | 0.1339 | 0.3083 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3311C>A | P1104Q 2D AIThe SynGAP1 missense variant P1104Q is reported in gnomAD (ID 6‑33443863‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign likelihood. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | 6-33443863-C-A | -3.161 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.64 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.68 | Benign | 0.06 | Tolerated | 3.77 | 5 | 0.1498 | 0.5063 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||
| c.3845A>G | E1282G 2D  AIThe SynGAP1 missense variant E1282G is reported in gnomAD (ID 6‑33447893‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Taken together, the overwhelming majority of predictions support a benign impact, and there is no ClinVar classification to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.817364 | Binding | 0.465 | 0.725 | 0.875 | 6-33447893-A-G | -2.649 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.219 | Likely Benign | -1.25 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.68 | Benign | 0.00 | Affected | 0.2729 | 0.5284 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||
| c.3850C>A | L1284M 2D AIThe SynGAP1 missense variant L1284M is reported in gnomAD (ID 6‑33447898‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from the four benign‑oriented tools). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.824557 | Binding | 0.441 | 0.748 | 0.875 | 6-33447898-C-A | -5.332 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.49 | Neutral | 0.970 | Probably Damaging | 0.637 | Possibly Damaging | 2.61 | Benign | 0.03 | Affected | 3.77 | 5 | 0.0731 | 0.2092 | 2 | 4 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||
| c.3902C>A | P1301H 2D AIThe SynGAP1 missense variant P1301H is listed in ClinVar (ID 212356.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33451776‑C‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus methods report a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No Foldetta stability result is available. Overall, the majority of predictions, including the high‑accuracy consensus, support a benign classification, which does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.885064 | Binding | 0.447 | 0.841 | 0.875 | Conflicting | 2 | 6-33451776-C-A | 5 | 3.10e-6 | -5.756 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.232 | Likely Benign | -1.13 | Neutral | 0.642 | Possibly Damaging | 0.378 | Benign | 2.79 | Benign | 0.04 | Affected | 3.77 | 5 | 0.1305 | 0.2965 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||
| c.3941C>A | P1314Q 2D AIThe SynGAP1 missense variant P1314Q is listed in gnomAD (ID 6‑33451815‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign. No Foldetta stability data are available, so folding‑stability evidence is unavailable. Overall, the preponderance of computational evidence indicates that P1314Q is most likely benign, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.971592 | Binding | 0.467 | 0.903 | 0.750 | 6-33451815-C-A | -4.222 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.55 | Neutral | 0.618 | Possibly Damaging | 0.333 | Benign | 4.24 | Benign | 0.04 | Affected | 3.77 | 5 | 0.1612 | 0.4588 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||
| c.44C>G | A15G 2D AIThe SynGAP1 missense variant A15G is reported in gnomAD (ID 6‑33420308‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus also reports likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.466055 | Uncertain | 0.330 | 0.912 | 0.375 | 6-33420308-C-G | 3 | 1.95e-6 | -3.261 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -0.04 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2753 | 0.5196 | 0 | 1 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.653T>A | F218Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in silico predictors classify the substitution as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign effect. The high‑accuracy folding‑stability tool Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign impact. No tool predicts pathogenicity, and no contradictory evidence exists in ClinVar. **Based on the consensus of all predictions, the variant is most likely benign, and this assessment does not conflict with ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | -3.596 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.26 | Likely Benign | 0.1 | -0.40 | Likely Benign | -0.07 | Likely Benign | -0.44 | Likely Benign | 0.229 | Likely Benign | 0.63 | Neutral | 0.001 | Benign | 0.002 | Benign | 5.95 | Benign | 0.52 | Tolerated | 0.1532 | 0.2725 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.778G>A | V260I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V260I missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts benign. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.254060 | Structured | 0.382651 | Uncertain | 0.888 | 0.259 | 0.250 | -7.525 | In-Between | 0.104 | Likely Benign | Likely Benign | -0.16 | Likely Benign | 0.1 | 0.27 | Likely Benign | 0.06 | Likely Benign | -0.14 | Likely Benign | 0.404 | Likely Benign | -0.85 | Neutral | 0.994 | Probably Damaging | 0.970 | Probably Damaging | 5.83 | Benign | 0.19 | Tolerated | 0.0559 | 0.3704 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.892C>A | P298T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P298T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar) and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain results and are treated as unavailable for pathogenicity inference. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the preponderance of evidence points to a benign effect for P298T, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -7.366 | In-Between | 0.104 | Likely Benign | Likely Benign | 1.44 | Ambiguous | 0.2 | 1.03 | Ambiguous | 1.24 | Ambiguous | 0.04 | Likely Benign | 0.209 | Likely Benign | -0.13 | Neutral | 0.939 | Possibly Damaging | 0.739 | Possibly Damaging | 1.96 | Pathogenic | 1.00 | Tolerated | 0.1595 | 0.6349 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.1148G>C | G383A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as pathogenic. Thus, the majority of evidence points to a benign impact, with only a minority of high‑accuracy tools suggesting pathogenicity. The variant is most likely benign based on the overall predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -6.205 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 3.17 | Destabilizing | 1.2 | 3.47 | Destabilizing | 3.32 | Destabilizing | 0.04 | Likely Benign | 0.178 | Likely Benign | -0.64 | Neutral | 0.055 | Benign | 0.037 | Benign | 4.20 | Benign | 0.11 | Tolerated | 0.3880 | 0.4361 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1204C>G | L402V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L402V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a benign outcome. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result and is therefore not considered evidence for pathogenicity. In summary, all available predictions support a benign classification for the L402V variant, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.243554 | Structured | 0.431978 | Uncertain | 0.961 | 0.383 | 0.000 | -3.467 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 1.91 | Ambiguous | 0.1 | 0.78 | Ambiguous | 1.35 | Ambiguous | -0.11 | Likely Benign | 0.203 | Likely Benign | 0.18 | Neutral | 0.004 | Benign | 0.004 | Benign | 4.01 | Benign | 0.29 | Tolerated | 0.1807 | 0.3657 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1312G>T | A438S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A438S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree that the substitution is benign: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as benign. The only inconclusive results come from Rosetta and premPS, which are listed as uncertain. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. Overall, the evidence strongly supports a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.147574 | Structured | 0.290154 | Uncertain | 0.929 | 0.293 | 0.000 | -6.085 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.30 | Likely Benign | 0.0 | 0.62 | Ambiguous | 0.46 | Likely Benign | 0.68 | Ambiguous | 0.012 | Likely Benign | -1.27 | Neutral | 0.042 | Benign | 0.035 | Benign | 4.14 | Benign | 0.15 | Tolerated | 0.2143 | 0.3995 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1720C>G | L574V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L574V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. No other high‑confidence pathogenic predictions are present. Based on the preponderance of benign predictions and the lack of any ClinVar pathogenic annotation, the variant is most likely benign. This conclusion does not contradict any existing ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | -5.559 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.78 | Ambiguous | 0.1 | 0.37 | Likely Benign | 0.58 | Ambiguous | 0.25 | Likely Benign | 0.149 | Likely Benign | -0.40 | Neutral | 0.004 | Benign | 0.003 | Benign | -1.27 | Pathogenic | 0.29 | Tolerated | 0.1481 | 0.2978 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2257G>C | A753P 2D AIThe SynGAP1 missense variant A753P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -2.486 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.05 | Neutral | 0.966 | Probably Damaging | 0.575 | Possibly Damaging | 2.59 | Benign | 0.30 | Tolerated | 0.2143 | 0.5442 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2405G>C | G802A 2D AIThe SynGAP1 missense variant G802A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Benign” status. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G802A is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.894241 | Disordered | 0.681966 | Binding | 0.294 | 0.898 | 0.625 | -3.584 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.57 | Neutral | 0.059 | Benign | 0.089 | Benign | 2.74 | Benign | 0.02 | Affected | 0.3779 | 0.4554 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2507G>C | S836T 2D AIThe SynGAP1 missense variant S836T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.634582 | Binding | 0.269 | 0.859 | 0.250 | -3.871 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -1.28 | Neutral | 0.901 | Possibly Damaging | 0.535 | Possibly Damaging | 2.56 | Benign | 0.36 | Tolerated | 0.1161 | 0.5326 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2638G>C | A880P 2D AIThe SynGAP1 missense variant A880P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.621441 | Binding | 0.309 | 0.874 | 0.250 | -3.671 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -0.55 | Neutral | 0.971 | Probably Damaging | 0.693 | Possibly Damaging | 2.61 | Benign | 0.11 | Tolerated | 0.1744 | 0.4106 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2648T>C | L883P 2D AIThe SynGAP1 missense variant L883P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence supports a benign classification for this variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.641952 | Binding | 0.334 | 0.886 | 0.250 | -2.572 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.58 | Neutral | 0.934 | Possibly Damaging | 0.516 | Possibly Damaging | 2.62 | Benign | 0.21 | Tolerated | 0.3462 | 0.1658 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2840G>T | G947V 2D AIThe SynGAP1 missense variant G947V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign, whereas polyPhen‑2 HumDiv and SIFT predict pathogenicity; ESM1b remains uncertain. High‑accuracy methods reinforce the benign assessment: AlphaMissense‑Optimized scores benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and Foldetta data are not available. Overall, the preponderance of evidence points to a benign impact for G947V, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | -7.171 | In-Between | 0.105 | Likely Benign | Likely Benign | 0.296 | Likely Benign | -1.11 | Neutral | 0.586 | Possibly Damaging | 0.303 | Benign | 4.93 | Benign | 0.01 | Affected | 0.1443 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.305T>C | L102S 2D  AIThe SynGAP1 missense variant L102S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus likewise indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for the L102S variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.696014 | Binding | 0.357 | 0.885 | 0.625 | -3.260 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.174 | Likely Benign | 0.54 | Neutral | 0.984 | Probably Damaging | 0.969 | Probably Damaging | 4.18 | Benign | 0.00 | Affected | 0.2785 | 0.1752 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3323G>C | S1108T 2D AIThe SynGAP1 missense variant S1108T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -5.710 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.49 | Neutral | 0.393 | Benign | 0.239 | Benign | 2.56 | Benign | 0.25 | Tolerated | 0.1304 | 0.5365 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3362G>A | S1121N 2D AIThe SynGAP1 missense variant S1121N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the consensus of the majority of predictors and the high‑accuracy tools points to a benign classification, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | -6.564 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.221 | Likely Benign | -0.12 | Neutral | 0.802 | Possibly Damaging | 0.266 | Benign | 5.50 | Benign | 0.00 | Affected | 0.1980 | 0.4211 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3412T>G | S1138A 2D AIThe SynGAP1 missense variant S1138A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.738250 | Binding | 0.346 | 0.869 | 1.000 | -5.821 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.354 | Likely Benign | -0.99 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 5.47 | Benign | 0.24 | Tolerated | 0.4387 | 0.5748 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3418A>T | T1140S 2D AIThe SynGAP1 missense variant T1140S is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.708094 | Binding | 0.293 | 0.854 | 1.000 | -2.805 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -0.27 | Neutral | 0.025 | Benign | 0.010 | Benign | 3.25 | Benign | 0.91 | Tolerated | 0.3401 | 0.3504 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.37A>G | I13V 2D AIThe SynGAP1 missense variant I13V is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the ClinVar designation of uncertain significance rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.482657 | Uncertain | 0.318 | 0.916 | 0.375 | Uncertain | 2 | -2.497 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.110 | Likely Benign | 0.01 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1548 | 0.4315 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.3881C>T | A1294V 2D AIThe SynGAP1 missense variant A1294V is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447929‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑benign/2‑pathogenic split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized as benign, SGM Consensus as inconclusive, and Foldetta as unavailable. Overall, the predictions are mixed, with one more pathogenic than benign calls. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447929-C-T | 1 | 6.45e-7 | -3.842 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -3.16 | Deleterious | 0.997 | Probably Damaging | 0.983 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1033 | 0.4413 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||
| c.4028A>G | H1343R 2D  AIThe SynGAP1 missense variant H1343R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.983646 | Binding | 0.350 | 0.677 | 0.875 | -2.179 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -1.16 | Neutral | 0.659 | Possibly Damaging | 0.071 | Benign | 4.08 | Benign | 0.00 | Affected | 0.2301 | 0.2602 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.613A>C | I205L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I205L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No contradictory evidence exists. Based on the collective predictions, the variant is most likely benign, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.264545 | Structured | 0.409933 | Uncertain | 0.821 | 0.414 | 0.125 | -5.474 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.07 | Likely Benign | 0.2 | 0.02 | Likely Benign | 0.05 | Likely Benign | 0.30 | Likely Benign | 0.112 | Likely Benign | -0.86 | Neutral | 0.004 | Benign | 0.012 | Benign | 4.17 | Benign | 0.34 | Tolerated | 0.0682 | 0.2702 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.905C>G | S302C 2D AIThe SynGAP1 missense variant S302C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, premPS, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Uncertain predictions come from Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for S302C, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.263489 | Uncertain | 0.616 | 0.258 | 0.375 | -7.290 | In-Between | 0.105 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.5 | 1.24 | Ambiguous | 0.78 | Ambiguous | -0.04 | Likely Benign | 0.070 | Likely Benign | -0.83 | Neutral | 0.833 | Possibly Damaging | 0.455 | Possibly Damaging | 4.05 | Benign | 0.02 | Affected | 0.1221 | 0.6514 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.908G>C | G303A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G303A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX, Rosetta, and Foldetta, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, while Foldetta’s stability analysis is uncertain. Overall, the variant is most likely benign based on the consensus of predictive tools, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.450668 | Structured | 0.271087 | Uncertain | 0.630 | 0.254 | 0.250 | -1.965 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 1.66 | Ambiguous | 0.2 | -0.51 | Ambiguous | 0.58 | Ambiguous | 0.10 | Likely Benign | 0.034 | Likely Benign | -0.90 | Neutral | 0.112 | Benign | 0.058 | Benign | 4.07 | Benign | 0.35 | Tolerated | 0.3724 | 0.5325 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1121C>G | S374C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S374C is reported in gnomAD (6-33438026-C-G) and has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar all indicate benign. Only two tools (polyPhen2_HumDiv and SIFT) predict pathogenicity, while the consensus score SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM‑Consensus itself is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. No prediction or stability result is missing or inconclusive. Based on the aggregate evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.428948 | Uncertain | 0.333 | 0.812 | 0.625 | 6-33438026-C-G | -6.242 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.79 | Ambiguous | 0.45 | Likely Benign | 0.08 | Likely Benign | 0.317 | Likely Benign | -0.99 | Neutral | 0.875 | Possibly Damaging | 0.430 | Benign | 5.30 | Benign | 0.00 | Affected | 4.32 | 13 | 0.1749 | 0.6584 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||
| c.1222A>T | T408S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T408S is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, FoldX, Rosetta, premPS, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool predicts pathogenicity. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates benign stability. Consequently, the variant is most likely benign, and this prediction does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | -6.277 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.39 | Likely Benign | 0.25 | Likely Benign | 0.18 | Likely Benign | 0.044 | Likely Benign | -1.48 | Neutral | 0.182 | Benign | 0.127 | Benign | 4.24 | Benign | 0.21 | Tolerated | 0.3256 | 0.4767 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.122G>A | R41H 2D AIThe SynGAP1 missense variant R41H is reported in gnomAD (variant ID 6-33423531‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: the majority (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a benign effect, while a minority (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT) predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized classifies the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Benign. No Foldetta stability data are available, so it does not influence the overall assessment. Based on the preponderance of evidence from both general and high‑accuracy predictors, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.311707 | Structured | 0.431757 | Uncertain | 0.344 | 0.765 | 0.375 | 6-33423531-G-A | 6 | 3.72e-6 | -4.425 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -0.74 | Neutral | 0.976 | Probably Damaging | 0.848 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3388 | 0.3065 | 0 | 2 | 1.3 | -19.05 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.2305C>G | L769V 2D AIThe SynGAP1 missense variant L769V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, but this is the sole discordant call. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -4.585 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.41 | Neutral | 0.625 | Possibly Damaging | 0.249 | Benign | 4.04 | Benign | 0.25 | Tolerated | 0.1364 | 0.2558 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2563C>T | L855F 2D AIThe SynGAP1 missense variant L855F is predicted to be benign by all evaluated in‑silico tools. Consensus predictions from **SGM‑Consensus** (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classify the variant as *Likely Benign*. High‑accuracy predictors **AlphaMissense‑Optimized** also report a benign effect. Other pathogenicity predictors—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default—uniformly predict benign. No tools predict pathogenicity. **Foldetta**, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its stability impact remains unknown. The variant is not listed in ClinVar and has no entry in gnomAD, so no population frequency or clinical annotation is available. Based on the unanimous benign predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.485558 | Uncertain | 0.285 | 0.823 | 0.625 | -4.985 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.86 | Neutral | 0.411 | Benign | 0.187 | Benign | 4.00 | Benign | 0.12 | Tolerated | 0.0700 | 0.3529 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2663C>T | A888V 2D AIThe SynGAP1 missense variant A888V is catalogued in gnomAD (ID 6‑33443215‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes, while the single pathogenic signal comes from SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign likelihood. Foldetta results are unavailable, so they do not influence the assessment. Overall, the preponderance of evidence points to a benign impact for A888V, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.575860 | Binding | 0.352 | 0.928 | 0.625 | 6-33443215-C-T | 1 | 6.20e-7 | -4.241 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -0.91 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.78 | Benign | 0.00 | Affected | 4.32 | 4 | 0.1285 | 0.7056 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.2737A>T | T913S 2D AIThe SynGAP1 missense variant T913S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are not available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.763517 | Binding | 0.339 | 0.899 | 0.375 | -2.636 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -0.74 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.80 | Benign | 0.74 | Tolerated | 0.3541 | 0.5147 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.281C>A | P94H 2D AIThe SynGAP1 missense variant P94H is reported in gnomAD (variant ID 6‑33425889‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that P94H is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.570978 | Binding | 0.350 | 0.869 | 0.625 | 6-33425889-C-A | 1 | 6.20e-7 | -3.708 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -2.31 | Neutral | 0.637 | Possibly Damaging | 0.102 | Benign | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1486 | 0.3913 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||
| c.3176G>T | G1059V 2D AIThe SynGAP1 missense variant G1059V is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while the majority‑vote SGM‑Consensus also classifies it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy tools corroborate the benign prediction: AlphaMissense‑Optimized is benign and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign; Foldetta results are not available. Overall, the preponderance of evidence supports a benign classification for G1059V, and this assessment does not conflict with ClinVar, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | -7.242 | In-Between | 0.106 | Likely Benign | Likely Benign | 0.478 | Likely Benign | -0.82 | Neutral | 0.259 | Benign | 0.066 | Benign | 2.54 | Benign | 0.00 | Affected | 0.1462 | 0.3494 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3182G>T | G1061V 2D AIThe SynGAP1 missense variant G1061V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that G1061V is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | -6.709 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.307 | Likely Benign | -1.41 | Neutral | 0.224 | Benign | 0.066 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1431 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3187G>T | G1063C 2D AIThe SynGAP1 missense variant G1063C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -8.315 | Likely Pathogenic | 0.106 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.07 | Neutral | 0.938 | Possibly Damaging | 0.477 | Possibly Damaging | 4.19 | Benign | 0.01 | Affected | 0.1440 | 0.4639 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3292A>T | S1098C 2D AIThe SynGAP1 missense variant S1098C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus likewise indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -6.553 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -1.46 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 2.65 | Benign | 0.12 | Tolerated | 0.1249 | 0.6233 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.32G>C | G11A 2D AIThe SynGAP1 missense variant G11A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | -3.611 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.28 | Neutral | 0.105 | Benign | 0.007 | Benign | 4.00 | Benign | 0.00 | Affected | 0.4045 | 0.5440 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3341G>A | S1114N 2D AIThe SynGAP1 missense variant S1114N is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), also reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | -6.089 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.51 | Neutral | 0.071 | Benign | 0.058 | Benign | 2.71 | Benign | 0.06 | Tolerated | 0.1416 | 0.4610 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3844G>C | E1282Q 2D  AIThe SynGAP1 missense variant E1282Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.817364 | Binding | 0.465 | 0.725 | 0.875 | -2.785 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.079 | Likely Benign | 1.05 | Neutral | 0.004 | Benign | 0.003 | Benign | 2.70 | Benign | 0.31 | Tolerated | 0.0965 | 0.5651 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.706G>T | A236S 2D AIThe SynGAP1 A236S variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include FoldX, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Predictions that are inconclusive or uncertain are Rosetta, Foldetta, premPS, and ESM1b. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for A236S, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.185198 | Structured | 0.329926 | Uncertain | 0.775 | 0.330 | 0.000 | -7.845 | In-Between | 0.106 | Likely Benign | Likely Benign | 0.40 | Likely Benign | 0.5 | 1.24 | Ambiguous | 0.82 | Ambiguous | 0.65 | Ambiguous | 0.648 | Likely Pathogenic | -2.30 | Neutral | 0.948 | Possibly Damaging | 0.588 | Possibly Damaging | 5.83 | Benign | 0.13 | Tolerated | 0.2641 | 0.5547 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1139G>C | G380A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G380A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FoldX predicts a pathogenic outcome, while Rosetta and Foldetta are benign or uncertain, respectively. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta as uncertain. Overall, the overwhelming majority of evidence points to a benign effect, with no conflict with ClinVar status (which has no entry for this variant). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -6.433 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 3.11 | Destabilizing | 1.0 | -0.15 | Likely Benign | 1.48 | Ambiguous | -0.06 | Likely Benign | 0.422 | Likely Benign | -0.53 | Neutral | 0.056 | Benign | 0.010 | Benign | 2.56 | Benign | 0.17 | Tolerated | 0.3915 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1289T>C | M430T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430T is reported in gnomAD (6‑33438194‑T‑C) and has no ClinVar entry. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, FoldX (uncertain), Rosetta, Foldetta (uncertain), premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as benign or likely benign. No tool predicts pathogenicity. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is benign, the SGM Consensus is likely benign, and Foldetta is uncertain. Overall, the computational evidence strongly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic report. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | 6-33438194-T-C | 1 | 6.19e-7 | -3.430 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 1.89 | Ambiguous | 0.1 | 0.01 | Likely Benign | 0.95 | Ambiguous | 0.43 | Likely Benign | 0.103 | Likely Benign | -1.48 | Neutral | 0.016 | Benign | 0.010 | Benign | 3.48 | Benign | 0.40 | Tolerated | 3.37 | 29 | 0.2002 | 0.3184 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||
| c.1298C>G | A433G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A433G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach a consensus all indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. The protein‑folding stability method Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result and is therefore treated as unavailable evidence. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | -5.044 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.59 | Ambiguous | 0.0 | 1.21 | Ambiguous | 0.90 | Ambiguous | 0.70 | Ambiguous | 0.038 | Likely Benign | -1.54 | Neutral | 0.035 | Benign | 0.014 | Benign | 3.38 | Benign | 0.15 | Tolerated | 0.1629 | 0.2919 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1919C>A | T640N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. Only the protein‑stability predictors FoldX and Rosetta returned uncertain results, which are treated as unavailable evidence. High‑accuracy assessments corroborate the benign prediction: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also reports a benign effect. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | -4.258 | Likely Benign | 0.107 | Likely Benign | Likely Benign | -0.50 | Ambiguous | 0.2 | 0.53 | Ambiguous | 0.02 | Likely Benign | 0.03 | Likely Benign | 0.097 | Likely Benign | 0.51 | Neutral | 0.229 | Benign | 0.084 | Benign | 3.45 | Benign | 0.25 | Tolerated | 0.1518 | 0.4079 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.2311T>G | S771A 2D AIThe SynGAP1 missense variant S771A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -4.337 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.09 | Neutral | 0.025 | Benign | 0.014 | Benign | 4.09 | Benign | 0.62 | Tolerated | 0.5201 | 0.4745 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.2554G>T | G852C 2D AIThe SynGAP1 missense variant G852C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and Foldetta results are unavailable. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign,” and AlphaMissense‑Optimized independently predicts a benign outcome. With the majority of evidence pointing to a benign effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.506063 | Binding | 0.276 | 0.816 | 0.625 | -7.462 | In-Between | 0.107 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -1.75 | Neutral | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 4.10 | Benign | 0.01 | Affected | 0.1278 | 0.4808 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2581T>C | S861P 2D AIThe SynGAP1 missense variant S861P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for S861P, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.540903 | Binding | 0.285 | 0.797 | 0.250 | -4.256 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -1.65 | Neutral | 0.960 | Probably Damaging | 0.761 | Possibly Damaging | 3.93 | Benign | 0.13 | Tolerated | 0.1830 | 0.6011 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2587C>G | L863V 2D AIThe SynGAP1 missense variant L863V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for the L863V variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.594839 | Binding | 0.267 | 0.795 | 0.250 | -3.651 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -0.72 | Neutral | 0.995 | Probably Damaging | 0.926 | Probably Damaging | 4.09 | Benign | 0.21 | Tolerated | 0.1655 | 0.3544 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2594C>G | A865G 2D AIThe SynGAP1 missense variant A865G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.626222 | Binding | 0.271 | 0.788 | 0.250 | -3.412 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -0.74 | Neutral | 0.009 | Benign | 0.019 | Benign | 2.69 | Benign | 0.51 | Tolerated | 0.2344 | 0.4683 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2984C>A | P995H 2D AIThe SynGAP1 missense variant P995H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P995H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -5.347 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.75 | Neutral | 0.832 | Possibly Damaging | 0.600 | Possibly Damaging | 4.16 | Benign | 0.00 | Affected | 0.1618 | 0.4571 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3200C>T | P1067L 2D AIThe SynGAP1 missense variant P1067L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of predictions and the consensus analysis indicate a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.461 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -3.01 | Deleterious | 0.951 | Possibly Damaging | 0.619 | Possibly Damaging | 2.76 | Benign | 0.01 | Affected | 0.2047 | 0.6198 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3299G>C | G1100A 2D AIThe SynGAP1 missense variant G1100A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.862 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.95 | Neutral | 0.943 | Possibly Damaging | 0.667 | Possibly Damaging | 2.01 | Pathogenic | 0.05 | Affected | 0.3442 | 0.5154 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3397A>C | I1133L 2D AIThe SynGAP1 missense variant I1133L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that I1133L is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -1.147 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.202 | Likely Benign | -0.26 | Neutral | 0.000 | Benign | 0.001 | Benign | 5.48 | Benign | 0.23 | Tolerated | 0.0911 | 0.4302 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3418A>G | T1140A 2D AIThe SynGAP1 missense variant T1140A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.708094 | Binding | 0.293 | 0.854 | 1.000 | -3.838 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -0.36 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.71 | Benign | 1.00 | Tolerated | 0.3949 | 0.3463 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3445C>G | P1149A 2D AIThe SynGAP1 missense variant P1149A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.786938 | Binding | 0.424 | 0.837 | 0.625 | -3.179 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.037 | Likely Benign | -0.72 | Neutral | 0.025 | Benign | 0.022 | Benign | 2.76 | Benign | 0.07 | Tolerated | 0.3403 | 0.4352 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.359G>T | G120V 2D AIThe SynGAP1 missense variant G120V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.659993 | Binding | 0.359 | 0.887 | 0.750 | -4.571 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -0.68 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.28 | Benign | 0.19 | Tolerated | 0.1321 | 0.3883 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3736A>C | K1246Q 2D AIThe SynGAP1 missense change K1246Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as “Likely Benign.” In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that K1246Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.750527 | Disordered | 0.375382 | Uncertain | 0.887 | 0.564 | 0.625 | -0.718 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.43 | Neutral | 0.961 | Probably Damaging | 0.721 | Possibly Damaging | 2.66 | Benign | 0.06 | Tolerated | 0.3708 | 0.0740 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||||
| c.55G>T | A19S 2D AIThe SynGAP1 missense variant A19S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | -2.905 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -0.21 | Neutral | 0.225 | Benign | 0.027 | Benign | 4.17 | Benign | 0.00 | Affected | 0.3273 | 0.6260 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.83C>G | S28C 2D AIThe SynGAP1 missense variant S28C is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.438157 | Uncertain | 0.354 | 0.884 | 0.125 | -4.800 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.021 | Likely Benign | -0.26 | Neutral | 0.022 | Benign | 0.004 | Benign | 4.13 | Benign | 0.11 | Tolerated | 0.1387 | 0.5741 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.893C>T | P298L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P298L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign outcome; and Foldetta’s stability prediction is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -7.334 | In-Between | 0.107 | Likely Benign | Likely Benign | 0.60 | Ambiguous | 0.2 | 1.53 | Ambiguous | 1.07 | Ambiguous | -0.16 | Likely Benign | 0.267 | Likely Benign | -0.82 | Neutral | 0.885 | Possibly Damaging | 0.589 | Possibly Damaging | 1.91 | Pathogenic | 0.21 | Tolerated | 0.2137 | 0.6795 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.913A>T | T305S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T305S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or uncertain are provided by Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.359901 | Structured | 0.299706 | Uncertain | 0.872 | 0.274 | 0.125 | -2.899 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.45 | Likely Benign | 0.4 | 1.21 | Ambiguous | 0.83 | Ambiguous | 0.55 | Ambiguous | 0.135 | Likely Benign | -0.60 | Neutral | 0.760 | Possibly Damaging | 0.484 | Possibly Damaging | 1.86 | Pathogenic | 0.54 | Tolerated | 0.3579 | 0.4496 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.914C>G | T305S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T305S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or uncertain are provided by Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.359901 | Structured | 0.299706 | Uncertain | 0.872 | 0.274 | 0.125 | -2.899 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.45 | Likely Benign | 0.4 | 1.21 | Ambiguous | 0.83 | Ambiguous | 0.55 | Ambiguous | 0.104 | Likely Benign | -0.60 | Neutral | 0.760 | Possibly Damaging | 0.484 | Possibly Damaging | 1.86 | Pathogenic | 0.54 | Tolerated | 0.3579 | 0.4496 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.967C>G | L323V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L323V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (REVEL, premPS, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and pathogenic predictions (FoldX, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM). The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled Likely Benign. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates Likely Benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts pathogenic. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.268042 | Structured | 0.428564 | Uncertain | 0.956 | 0.369 | 0.000 | -3.483 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 2.18 | Destabilizing | 0.3 | 2.22 | Destabilizing | 2.20 | Destabilizing | 0.17 | Likely Benign | 0.227 | Likely Benign | 0.19 | Neutral | 0.898 | Possibly Damaging | 0.472 | Possibly Damaging | 0.80 | Pathogenic | 0.80 | Tolerated | 0.1277 | 0.2656 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.103G>C | V35L 2D AIThe SynGAP1 missense variant V35L is reported in gnomAD (variant ID 6-33423512‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence, including the high‑accuracy tools, points to a benign effect. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | 6-33423512-G-C | 4 | 2.48e-6 | -2.893 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.58 | Neutral | 0.481 | Possibly Damaging | 0.506 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1026 | 0.4025 | 1 | 2 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||||
| c.2125C>A | L709M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -5.242 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.18 | Likely Benign | 0.0 | 0.40 | Likely Benign | 0.29 | Likely Benign | -0.10 | Likely Benign | 0.065 | Likely Benign | 0.03 | Neutral | 0.688 | Possibly Damaging | 0.235 | Benign | 3.41 | Benign | 0.17 | Tolerated | 0.0629 | 0.2409 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.2231A>G | Q744R 2D  AIThe SynGAP1 missense variant Q744R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -2.207 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.43 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.80 | Benign | 0.05 | Affected | 0.1437 | 0.1588 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2321C>T | A774V 2D AIThe SynGAP1 missense variant A774V is catalogued in gnomAD (ID 6‑33442479‑C‑T) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently scores the variant as benign. No pathogenic predictions are reported. Grouping by agreement, the benign‑predicting tools comprise the entire set, while the pathogenic group is empty. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | 6-33442479-C-T | 1 | 1.28e-6 | -3.075 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.73 | Neutral | 0.000 | Benign | 0.003 | Benign | 4.20 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1070 | 0.6659 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.2396C>A | P799H 2D AIThe SynGAP1 missense variant P799H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree that the substitution is benign: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign effect. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign classification for P799H, and this conclusion does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.537892 | Binding | 0.400 | 0.894 | 0.750 | -5.611 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.80 | Neutral | 0.999 | Probably Damaging | 0.933 | Probably Damaging | 4.20 | Benign | 0.00 | Affected | 0.1684 | 0.4192 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||
| c.2572A>T | S858C 2D AIThe SynGAP1 missense variant S858C is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.482724 | Uncertain | 0.305 | 0.833 | 0.375 | -6.767 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -1.93 | Neutral | 0.940 | Possibly Damaging | 0.979 | Probably Damaging | 4.06 | Benign | 0.02 | Affected | 0.1206 | 0.6155 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2747T>C | V916A 2D AIThe SynGAP1 missense variant V916A is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool in the dataset indicates a pathogenic effect, so the pathogenic‑prediction group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign,” and the Foldetta stability analysis is not available. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -2.524 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.18 | Neutral | 0.244 | Benign | 0.171 | Benign | 2.91 | Benign | 1.00 | Tolerated | 0.3455 | 0.2841 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2827G>T | G943C 2D AIThe SynGAP1 missense variant G943C is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b all predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy tools—points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | -9.822 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.356 | Likely Benign | -0.94 | Neutral | 0.938 | Possibly Damaging | 0.649 | Possibly Damaging | 2.84 | Benign | 0.10 | Tolerated | 0.1425 | 0.4439 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2867C>G | S956C 2D AIThe SynGAP1 missense variant S956C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. four benign) indicate a likely pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant has no existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -9.292 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -0.34 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 1.94 | Pathogenic | 0.03 | Affected | 0.1833 | 0.5470 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.2879A>T | H960L 2D AIThe SynGAP1 missense variant H960L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987911 | Disordered | 0.983385 | Binding | 0.380 | 0.901 | 0.750 | -8.386 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.30 | Neutral | 0.174 | Benign | 0.043 | Benign | 4.17 | Benign | 0.33 | Tolerated | 0.1465 | 0.5595 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2885A>T | H962L 2D AIThe SynGAP1 missense variant H962L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for H962L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.984483 | Binding | 0.369 | 0.886 | 0.750 | -8.478 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -1.49 | Neutral | 0.494 | Possibly Damaging | 0.170 | Benign | 4.15 | Benign | 0.03 | Affected | 0.1738 | 0.5487 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3149G>T | G1050V 2D AIThe SynGAP1 missense variant G1050V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for G1050V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -6.450 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.49 | Pathogenic | 0.13 | Tolerated | 0.1260 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3238G>T | A1080S 2D AIThe SynGAP1 missense variant A1080S is listed in ClinVar (ID 2703014.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443790‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not contradict the ClinVar designation, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | Uncertain | 1 | 6-33443790-G-T | 1 | 6.26e-7 | -3.277 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.103 | Likely Benign | 0.01 | Neutral | 0.702 | Possibly Damaging | 0.346 | Benign | 4.16 | Benign | 0.08 | Tolerated | 3.77 | 5 | 0.2498 | 0.5915 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||
| c.341A>G | K114R 2D AIThe SynGAP1 missense variant K114R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect; there is no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.649749 | Binding | 0.381 | 0.879 | 0.750 | -1.861 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.02 | Neutral | 0.005 | Benign | 0.003 | Benign | 4.09 | Benign | 0.00 | Affected | 0.5668 | 0.1498 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.357G>C | E119D 2D AIThe SynGAP1 missense variant E119D is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.661946 | Binding | 0.346 | 0.881 | 0.750 | -3.258 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.07 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.08 | Benign | 0.32 | Tolerated | 3.61 | 5 | 0.2180 | 0.5759 | 2 | 3 | 0.0 | -14.03 | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||||||||||||||||||||||
| c.357G>T | E119D 2D AIThe SynGAP1 missense variant E119D is reported in gnomAD (variant ID 6‑33432222‑G‑T) but has no ClinVar entry. All available in silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized indicates a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.661946 | Binding | 0.346 | 0.881 | 0.750 | 6-33432222-G-T | 3 | 1.86e-6 | -3.258 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.07 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.08 | Benign | 0.32 | Tolerated | 3.61 | 5 | 0.2180 | 0.5759 | 2 | 3 | 0.0 | -14.03 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.368C>T | A123V 2D AIThe SynGAP1 missense variant A123V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.689505 | Binding | 0.324 | 0.886 | 0.750 | -4.119 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.040 | Likely Benign | -0.77 | Neutral | 0.010 | Benign | 0.003 | Benign | 4.15 | Benign | 0.02 | Affected | 0.1549 | 0.6874 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.88C>A | H30N 2D AIThe SynGAP1 missense variant H30N is not reported in ClinVar and is absent from gnomAD. Consensus among most in silico predictors is benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect, while only SIFT predicts pathogenicity. Grouping the tools, the benign‑predicting set (nine tools) overwhelmingly outweighs the single pathogenic prediction. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the computational evidence strongly supports a benign classification, and this is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.570702 | Disordered | 0.438063 | Uncertain | 0.373 | 0.883 | 0.250 | -3.096 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -1.91 | Neutral | 0.273 | Benign | 0.380 | Benign | 3.92 | Benign | 0.00 | Affected | 0.2699 | 0.3818 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.899C>G | S300C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S300C is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33437804‑C‑G). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Rosetta and Foldetta give uncertain results and are therefore not considered evidence for either side. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the majority of reliable predictions indicate a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.256848 | Uncertain | 0.742 | 0.280 | 0.375 | 6-33437804-C-G | -6.749 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.2 | 1.45 | Ambiguous | 0.88 | Ambiguous | 0.34 | Likely Benign | 0.129 | Likely Benign | -2.45 | Neutral | 0.975 | Probably Damaging | 0.815 | Possibly Damaging | 1.55 | Pathogenic | 0.01 | Affected | 3.47 | 19 | 0.1005 | 0.6493 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||
| c.1022G>C | G341A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G341A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come only from polyPhen‑2 HumDiv and FATHMM. Uncertain results are reported by Rosetta and Foldetta. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is likely benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains inconclusive. Overall, the preponderance of evidence indicates that G341A is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -3.211 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.4 | -1.23 | Ambiguous | -0.54 | Ambiguous | -0.03 | Likely Benign | 0.239 | Likely Benign | -1.13 | Neutral | 0.625 | Possibly Damaging | 0.192 | Benign | 0.43 | Pathogenic | 0.15 | Tolerated | 0.3562 | 0.3979 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.104T>G | V35G 2D  AIThe SynGAP1 missense variant V35G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus also predicts benign; Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because the variant is not currently catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -3.614 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.143 | Likely Benign | 0.66 | Neutral | 0.693 | Possibly Damaging | 0.824 | Possibly Damaging | 4.41 | Benign | 0.00 | Affected | 0.1514 | 0.2618 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1145G>C | G382A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G382A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact. Overall, the majority of predictions lean toward a benign effect, and this consensus does not contradict any ClinVar status (none available). Thus, based on the available computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -6.323 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 2.90 | Destabilizing | 0.6 | 3.00 | Destabilizing | 2.95 | Destabilizing | -0.03 | Likely Benign | 0.495 | Likely Benign | -0.59 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 1.33 | Pathogenic | 0.12 | Tolerated | 0.4048 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.124C>T | P42S 2D AIThe SynGAP1 missense variant P42S is listed in gnomAD (ID 6‑33423533‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for P42S, and this conclusion is not contradicted by ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | 6-33423533-C-T | 1 | 6.20e-7 | -3.472 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.12 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3462 | 0.4470 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.127G>T | G43C 2D AIThe SynGAP1 missense variant G43C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -4.599 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.90 | Neutral | 0.987 | Probably Damaging | 0.750 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.1445 | 0.3438 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2014A>G | T672A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T672A is listed in ClinVar as Benign (ClinVar ID 2154412.0) and is present in gnomAD (variant ID 6‑33441273‑A‑G). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only PROVEAN predicts a pathogenic outcome. Uncertain results are reported for FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a benign effect, and this conclusion is consistent with the ClinVar designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | Benign | 1 | 6-33441273-A-G | 3 | 1.86e-6 | -6.524 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.51 | Ambiguous | 0.3 | 1.15 | Ambiguous | 0.83 | Ambiguous | 0.65 | Ambiguous | 0.046 | Likely Benign | -3.20 | Deleterious | 0.006 | Benign | 0.002 | Benign | 3.44 | Benign | 0.12 | Tolerated | 3.40 | 25 | 0.3687 | 0.4380 | 1 | 0 | 2.5 | -30.03 | 188.5 | 42.5 | -0.1 | 0.3 | 0.2 | 0.0 | X | Potentially Pathogenic | The hydroxyl group of Thr672, located in an entangled α-α loop connecting the two α-helices (res. Ser641-Glu666 and res. Leu685-Val699), is involved in a highly coordinated hydrogen-bonding network between residues from two α-helices (res. Ser641-Glu666 and res. Arg563-Glu578) and from the α-α loop itself, such as Lys566, Glu666, and Asn669. In the variant simulations, Ala672 can only form a hydrogen bond with Lys566 via its backbone carbonyl group. Consequently, it cannot maintain the Lys566-Glu666 salt bridge through hydrogen bonding, leading to a significant disruption of the intricate and stable hydrogen-bond network between the loop and the helices. | |||||||||||||
| c.2020A>C | T674P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T674P missense change is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The high‑accuracy AlphaMissense‑Optimized predicts benign, while Foldetta predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. No other high‑accuracy tool provides a definitive call. Consequently, the evidence is evenly split between benign and pathogenic, leaving the variant’s clinical significance uncertain. This uncertainty does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -8.661 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.91 | Ambiguous | 0.6 | 4.12 | Destabilizing | 2.52 | Destabilizing | 0.12 | Likely Benign | 0.143 | Likely Benign | -2.69 | Deleterious | 0.995 | Probably Damaging | 0.891 | Possibly Damaging | 3.50 | Benign | 0.16 | Tolerated | 0.2039 | 0.6103 | 0 | -1 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||
| c.2209C>G | Q737E 2D AIThe SynGAP1 missense variant Q737E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -5.288 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -1.05 | Neutral | 0.906 | Possibly Damaging | 0.629 | Possibly Damaging | 2.76 | Benign | 0.04 | Affected | 0.1635 | 0.2355 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2211G>C | Q737H 2D  AIThe SynGAP1 missense variant Q737H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect for Q737H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -4.517 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -1.55 | Neutral | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 2.72 | Benign | 0.04 | Affected | 0.1683 | 0.3707 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2211G>T | Q737H 2D AIThe SynGAP1 missense variant Q737H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q737H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -4.517 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -1.55 | Neutral | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 2.72 | Benign | 0.04 | Affected | 0.1683 | 0.3707 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2251C>T | P751S 2D AIThe SynGAP1 missense variant P751S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions indicate that P751S is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -4.157 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.80 | Neutral | 0.514 | Possibly Damaging | 0.216 | Benign | 2.70 | Benign | 0.33 | Tolerated | 0.3446 | 0.5837 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2278A>G | M760V 2D AIThe SynGAP1 missense variant M760V is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence strongly supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.803 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.20 | Neutral | 0.001 | Benign | 0.008 | Benign | 2.69 | Benign | 0.22 | Tolerated | 0.3974 | 0.4381 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2569A>T | S857C 2D AIThe SynGAP1 missense variant S857C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the majority of predictions and the high‑accuracy consensus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.475747 | Uncertain | 0.288 | 0.826 | 0.375 | -6.335 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -1.28 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 4.02 | Benign | 0.08 | Tolerated | 0.1221 | 0.6672 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2837G>T | G946V 2D AIThe SynGAP1 missense variant G946V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G946V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985417 | Disordered | 0.845792 | Binding | 0.357 | 0.920 | 0.750 | -7.528 | In-Between | 0.109 | Likely Benign | Likely Benign | 0.368 | Likely Benign | -0.30 | Neutral | 0.901 | Possibly Damaging | 0.516 | Possibly Damaging | 4.57 | Benign | 0.00 | Affected | 0.1524 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2882A>T | H961L 2D AIThe SynGAP1 missense variant H961L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT and ESM1b predict pathogenicity, but these are outliers among the consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, SGM‑Consensus is benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.989835 | Disordered | 0.984562 | Binding | 0.323 | 0.893 | 0.750 | -8.547 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.155 | Likely Benign | -1.21 | Neutral | 0.144 | Benign | 0.078 | Benign | 4.13 | Benign | 0.01 | Affected | 0.1465 | 0.5344 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2888A>T | H963L 2D AIThe SynGAP1 missense variant H963L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates benign, and the SGM‑Consensus likewise suggests a benign effect; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for H963L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.983973 | Binding | 0.325 | 0.886 | 0.750 | -8.110 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -1.58 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.13 | Benign | 0.43 | Tolerated | 0.1626 | 0.5680 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2922C>A | D974E 2D AIThe SynGAP1 missense variant D974E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.617 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.63 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.26 | Benign | 0.05 | Affected | 0.2119 | 0.7516 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2922C>G | D974E 2D AIThe SynGAP1 missense variant D974E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.617 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.63 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.26 | Benign | 0.05 | Affected | 0.2119 | 0.7516 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2969C>G | S990C 2D AIThe SynGAP1 missense variant S990C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for S990C, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -5.753 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -1.91 | Neutral | 0.938 | Possibly Damaging | 0.690 | Possibly Damaging | 2.73 | Benign | 0.01 | Affected | 0.1041 | 0.5823 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.298T>A | Y100N 2D  AIThe SynGAP1 missense variant Y100N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -2.579 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -0.66 | Neutral | 0.675 | Possibly Damaging | 0.099 | Benign | 4.23 | Benign | 0.00 | Affected | 0.2487 | 0.0373 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3241G>T | A1081S 2D AIThe SynGAP1 missense variant A1081S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -3.536 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.29 | Neutral | 0.021 | Benign | 0.031 | Benign | 4.02 | Benign | 0.23 | Tolerated | 0.2268 | 0.4915 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3296A>G | Y1099C 2D  AIThe SynGAP1 missense variant Y1099C is reported in gnomAD (variant ID 6‑33443848‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | 6-33443848-A-G | 3 | 1.95e-6 | -5.670 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.13 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 2.73 | Benign | 0.14 | Tolerated | 3.77 | 5 | 0.3008 | 0.2382 | -2 | 0 | 3.8 | -60.04 | ||||||||||||||||||||||||||||||||||
| c.3325C>T | L1109F 2D AIThe SynGAP1 missense variant L1109F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -3.459 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -1.04 | Neutral | 0.832 | Possibly Damaging | 0.324 | Benign | 2.74 | Benign | 0.12 | Tolerated | 0.0780 | 0.4540 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3607C>A | H1203N 2D AIThe SynGAP1 missense variant H1203N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this view: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign, and Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.618285 | Disordered | 0.527023 | Binding | 0.892 | 0.589 | 0.250 | -4.278 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.181 | Likely Benign | -1.07 | Neutral | 0.002 | Benign | 0.018 | Benign | 5.61 | Benign | 0.31 | Tolerated | 0.1155 | 0.0914 | 2 | 1 | -0.3 | -23.04 | ||||||||||||||||||||||||||||||||||||||
| c.3802C>G | L1268V 2D AIThe SynGAP1 missense variant L1268V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports likely benign. Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for the variant, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.458154 | Structured | 0.804315 | Binding | 0.859 | 0.629 | 0.000 | -4.137 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.24 | Neutral | 0.649 | Possibly Damaging | 0.157 | Benign | 2.73 | Benign | 0.25 | Tolerated | 0.1331 | 0.2289 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3899C>T | P1300L 2D AIThe SynGAP1 missense variant P1300L is reported in gnomAD (variant ID 6‑33451773‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of tools, including the high‑accuracy methods, supports a benign interpretation. This prediction does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.885826 | Binding | 0.400 | 0.834 | 0.875 | 6-33451773-C-T | 1 | 6.20e-7 | -3.562 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -1.35 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.84 | Benign | 0.19 | Tolerated | 3.77 | 5 | 0.2348 | 0.5680 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3935C>T | A1312V 2D AIThe SynGAP1 missense variant A1312V is reported in gnomAD (ID 6‑33451809‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: six methods (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score all predict a benign effect, while three tools (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT) predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns “Benign” and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No Foldetta stability data are available, so it does not influence the conclusion. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status (none is listed). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.767246 | Disordered | 0.971112 | Binding | 0.392 | 0.902 | 0.750 | 6-33451809-C-T | 4 | 2.48e-6 | -4.585 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.231 | Likely Benign | -1.74 | Neutral | 0.991 | Probably Damaging | 0.983 | Probably Damaging | 3.15 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1495 | 0.6343 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.955G>C | A319P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319P is catalogued in gnomAD (ID 6‑33437860‑G‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive are Rosetta, ESM1b, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | 6-33437860-G-C | 3 | 1.86e-6 | -7.213 | In-Between | 0.109 | Likely Benign | Likely Benign | -0.02 | Likely Benign | 0.9 | -1.18 | Ambiguous | -0.60 | Ambiguous | 0.22 | Likely Benign | 0.286 | Likely Benign | 0.11 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 1.97 | Pathogenic | 1.00 | Tolerated | 3.38 | 23 | 0.1839 | 0.5067 | -1 | 1 | -3.4 | 26.04 | |||||||||||||||||||||||||
| c.1163G>C | G388A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G388A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as Pathogenic. Overall, the predictions are mixed, but the two most reliable tools (AlphaMissense‑Optimized and SGM‑Consensus) favor a benign interpretation, while Foldetta suggests instability. Based on the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | -6.577 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 2.55 | Destabilizing | 1.9 | 3.57 | Destabilizing | 3.06 | Destabilizing | -0.04 | Likely Benign | 0.457 | Likely Benign | -0.57 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 1.33 | Pathogenic | 0.05 | Affected | 0.3969 | 0.4837 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1913A>G | K638R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). In contrast, PROVEAN and polyPhen‑2 HumDiv predict a pathogenic impact, while premPS remains inconclusive. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is benign. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | Uncertain | 1 | -2.700 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.1 | -0.04 | Likely Benign | 0.03 | Likely Benign | 0.53 | Ambiguous | 0.216 | Likely Benign | -2.55 | Deleterious | 0.649 | Possibly Damaging | 0.240 | Benign | 3.41 | Benign | 0.13 | Tolerated | 3.37 | 31 | 0.4026 | 0.0975 | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||
| c.19T>A | S7T 2D AIThe SynGAP1 missense variant S7T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -4.182 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -0.26 | Neutral | 0.024 | Benign | 0.007 | Benign | 4.13 | Benign | 0.00 | Affected | 0.1688 | 0.5919 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2190C>G | I730M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs)—all uniformly indicate a benign impact. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -3.149 | Likely Benign | 0.110 | Likely Benign | Likely Benign | -0.15 | Likely Benign | 0.1 | 0.31 | Likely Benign | 0.08 | Likely Benign | -0.15 | Likely Benign | 0.042 | Likely Benign | -0.77 | Neutral | 0.993 | Probably Damaging | 0.914 | Probably Damaging | 3.43 | Benign | 0.09 | Tolerated | 0.0698 | 0.2159 | 2 | 1 | -2.6 | 18.03 | ||||||||||||||||||||||||||||||
| c.2192A>T | Q731L 2D AIThe SynGAP1 missense variant Q731L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign impact for Q731L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -4.251 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -1.27 | Neutral | 0.825 | Possibly Damaging | 0.270 | Benign | 2.75 | Benign | 0.12 | Tolerated | 0.0879 | 0.5694 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2305C>A | L769I 2D AIThe SynGAP1 missense variant L769I is listed in gnomAD (ID 6‑33442463‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | 6-33442463-C-A | -3.993 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.15 | Neutral | 0.836 | Possibly Damaging | 0.329 | Benign | 4.09 | Benign | 0.04 | Affected | 3.64 | 6 | 0.0800 | 0.3108 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||
| c.235A>C | N79H 2D AIThe SynGAP1 missense variant N79H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -2.090 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.72 | Neutral | 0.939 | Possibly Damaging | 0.114 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1247 | 0.4718 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3038C>G | S1013C 2D AIThe SynGAP1 missense variant S1013C is listed in ClinVar with an uncertain significance (ClinVar ID 934570.0) and is present in gnomAD (ID 6‑33443590‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.899570 | Binding | 0.308 | 0.846 | 0.625 | Uncertain | 1 | 6-33443590-C-G | 4 | 2.48e-6 | -6.745 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -2.06 | Neutral | 0.898 | Possibly Damaging | 0.579 | Possibly Damaging | 2.64 | Benign | 0.05 | Affected | 3.77 | 5 | 0.1345 | 0.5817 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||
| c.3044C>A | T1015N 2D AIThe SynGAP1 missense variant T1015N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.928486 | Binding | 0.295 | 0.823 | 0.625 | -4.353 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.027 | Likely Benign | 0.28 | Neutral | 0.004 | Benign | 0.002 | Benign | 2.54 | Benign | 0.24 | Tolerated | 0.1688 | 0.4233 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3161G>T | G1054V 2D AIThe SynGAP1 missense variant G1054V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -6.994 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -0.22 | Neutral | 0.818 | Possibly Damaging | 0.221 | Benign | 4.01 | Benign | 0.18 | Tolerated | 0.1578 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3364G>T | G1122C 2D AIThe SynGAP1 missense variant G1122C is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6‑33443916‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence—including the high‑confidence tools—supports a benign classification, and this conclusion does not contradict the ClinVar status, which currently has no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | 6-33443916-G-T | -8.895 | Likely Pathogenic | 0.110 | Likely Benign | Likely Benign | 0.375 | Likely Benign | -1.30 | Neutral | 0.975 | Probably Damaging | 0.733 | Possibly Damaging | 4.48 | Benign | 0.08 | Tolerated | 3.77 | 5 | 0.1343 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||
| c.3411T>A | H1137Q 2D AIThe SynGAP1 missense variant H1137Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while polyPhen‑2 (HumDiv and HumVar) and SIFT predict pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the pathogenic ones. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized scores benign, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.756488 | Binding | 0.314 | 0.879 | 0.875 | -2.958 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.239 | Likely Benign | -1.19 | Neutral | 0.925 | Possibly Damaging | 0.703 | Possibly Damaging | 5.34 | Benign | 0.00 | Affected | 0.1873 | 0.4090 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3411T>G | H1137Q 2D AIThe SynGAP1 missense variant H1137Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for H1137Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.756488 | Binding | 0.314 | 0.879 | 0.875 | -2.958 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.239 | Likely Benign | -1.19 | Neutral | 0.925 | Possibly Damaging | 0.703 | Possibly Damaging | 5.34 | Benign | 0.00 | Affected | 0.1873 | 0.4090 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.898T>C | S300P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S300P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The remaining tools—FoldX, Foldetta, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.356642 | Structured | 0.256848 | Uncertain | 0.742 | 0.280 | 0.375 | -7.379 | In-Between | 0.110 | Likely Benign | Likely Benign | 1.24 | Ambiguous | 1.0 | 0.06 | Likely Benign | 0.65 | Ambiguous | 0.50 | Likely Benign | 0.086 | Likely Benign | -2.46 | Neutral | 0.784 | Possibly Damaging | 0.390 | Benign | 1.54 | Pathogenic | 0.02 | Affected | 0.2192 | 0.5585 | 1 | -1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||
| c.97C>G | Q33E 2D AIThe SynGAP1 missense variant Q33E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign status: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.436712 | Uncertain | 0.342 | 0.860 | 0.375 | 0.191 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.024 | Likely Benign | -0.13 | Neutral | 0.017 | Benign | 0.014 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1773 | 0.3326 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.1099C>A | L367M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L367M variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and FATHMM. The remaining predictions are uncertain: Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also likely benign; Foldetta remains inconclusive. Overall, the majority of evidence supports a benign classification, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -4.968 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.1 | 0.92 | Ambiguous | 0.57 | Ambiguous | -0.04 | Likely Benign | 0.078 | Likely Benign | 0.12 | Neutral | 0.947 | Possibly Damaging | 0.360 | Benign | 1.63 | Pathogenic | 0.13 | Tolerated | 0.1402 | 0.3947 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.10T>G | S4A 2D AIThe SynGAP1 missense variant S4A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S4A, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.245 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.050 | Likely Benign | 0.02 | Neutral | 0.140 | Benign | 0.097 | Benign | 4.22 | Benign | 0.00 | Affected | 0.4871 | 0.5755 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.1133G>C | G378A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G378A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as “Likely Benign.” High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus confirms a benign likelihood, while Foldetta—combining FoldX‑MD and Rosetta outputs—predicts a pathogenic effect on protein folding stability. Overall, the majority of evidence points toward a benign impact, and this conclusion is consistent with the absence of ClinVar annotation and gnomAD data. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -6.450 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 5.06 | Destabilizing | 1.3 | 6.00 | Destabilizing | 5.53 | Destabilizing | -0.04 | Likely Benign | 0.497 | Likely Benign | -0.55 | Neutral | 0.999 | Probably Damaging | 0.981 | Probably Damaging | 1.33 | Pathogenic | 0.18 | Tolerated | 0.4062 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1229G>A | S410N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410N is predicted to be benign by all evaluated in‑silico tools. Consensus predictors (REVEL, SIFT, polyPhen‑2 HumDiv/HumVar, PROVEAN, premPS, FoldX, Rosetta, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly report a benign effect, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies it as “Likely Benign.” High‑accuracy assessments likewise support a benign outcome: AlphaMissense‑Optimized is benign, the SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) indicates a benign effect. ClinVar contains no entry for this variant, and it is not present in gnomAD. Based on the collective predictions, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -2.901 | Likely Benign | 0.111 | Likely Benign | Likely Benign | -0.16 | Likely Benign | 0.1 | -0.16 | Likely Benign | -0.16 | Likely Benign | 0.38 | Likely Benign | 0.049 | Likely Benign | -0.91 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.20 | Benign | 0.18 | Tolerated | 0.1310 | 0.5471 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||
| c.1831A>C | M611L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome, while Rosetta, Foldetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -6.721 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.1 | 0.65 | Ambiguous | 0.52 | Ambiguous | 0.56 | Ambiguous | 0.229 | Likely Benign | -1.29 | Neutral | 0.059 | Benign | 0.017 | Benign | -1.07 | Pathogenic | 0.76 | Tolerated | 0.1130 | 0.3547 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1831A>T | M611L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts pathogenic, while Rosetta, Foldetta, and premPS are uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates Likely Benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the preponderance of evidence points to a benign impact for M611L, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -6.721 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.1 | 0.65 | Ambiguous | 0.52 | Ambiguous | 0.56 | Ambiguous | 0.229 | Likely Benign | -1.29 | Neutral | 0.059 | Benign | 0.017 | Benign | -1.07 | Pathogenic | 0.76 | Tolerated | 0.1130 | 0.3547 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.19T>C | S7P 2D AIThe SynGAP1 missense variant S7P is reported in gnomAD (ID 6‑33420283‑T‑C) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign effect. This prediction is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | 6-33420283-T-C | -3.798 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.210 | Likely Benign | -0.23 | Neutral | 0.267 | Benign | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2474 | 0.4925 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||||
| c.2030G>A | S677N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677N is catalogued in gnomAD (6‑33441289‑G‑A) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while premPS and ESM1b are uncertain. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is benign. Thus, all available evidence supports a benign classification, and there is no conflict with ClinVar status (which is absent). The variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | 6-33441289-G-A | 1 | 6.20e-7 | -7.955 | In-Between | 0.111 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.5 | -0.15 | Likely Benign | 0.01 | Likely Benign | 0.89 | Ambiguous | 0.079 | Likely Benign | -2.05 | Neutral | 0.038 | Benign | 0.007 | Benign | 3.28 | Benign | 0.15 | Tolerated | 3.41 | 23 | 0.1763 | 0.5688 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||
| c.2335A>G | S779G 2D AIThe SynGAP1 missense variant S779G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.304 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.103 | Likely Benign | 0.38 | Neutral | 0.393 | Benign | 0.324 | Benign | 2.65 | Benign | 0.53 | Tolerated | 0.2894 | 0.5087 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2624C>G | A875G 2D AIThe SynGAP1 missense variant A875G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.632173 | Binding | 0.273 | 0.872 | 0.250 | -2.904 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.22 | Neutral | 0.022 | Benign | 0.048 | Benign | 2.68 | Benign | 0.04 | Affected | 0.2308 | 0.5046 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2734A>C | T912P 2D AIThe SynGAP1 missense variant T912P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.740671 | Binding | 0.285 | 0.909 | 0.250 | -2.955 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -0.78 | Neutral | 0.217 | Benign | 0.121 | Benign | 4.00 | Benign | 0.00 | Affected | 0.1832 | 0.4608 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.281C>T | P94L 2D AIThe SynGAP1 missense variant P94L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.570978 | Binding | 0.350 | 0.869 | 0.625 | -2.721 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -2.27 | Neutral | 0.198 | Benign | 0.017 | Benign | 4.13 | Benign | 0.00 | Affected | 0.2125 | 0.5862 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.4029C>A | H1343Q 2D AIThe SynGAP1 missense variant H1343Q is reported in gnomAD (ID 6‑33451903‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.983646 | Binding | 0.350 | 0.677 | 0.875 | 6-33451903-C-A | -2.900 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -1.04 | Neutral | 0.659 | Possibly Damaging | 0.104 | Benign | 4.06 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2126 | 0.3716 | 0 | 3 | -0.3 | -9.01 | ||||||||||||||||||||||||||||||||||||
| c.4029C>G | H1343Q 2D AIThe SynGAP1 missense variant H1343Q is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are polyPhen2_HumDiv and SIFT. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑accuracy tools, suggest that H1343Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.983646 | Binding | 0.350 | 0.677 | 0.875 | -2.900 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -1.04 | Neutral | 0.659 | Possibly Damaging | 0.104 | Benign | 4.06 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2126 | 0.3716 | 0 | 3 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||
| c.958G>A | V320I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V320I is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while Rosetta remains inconclusive. High‑accuracy assessments corroborate the benign trend: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the preponderance of evidence points to a benign effect for V320I, and this conclusion does not conflict with the current ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.185198 | Structured | 0.419626 | Uncertain | 0.905 | 0.266 | 0.125 | Uncertain | 1 | -5.220 | Likely Benign | 0.111 | Likely Benign | Likely Benign | -0.27 | Likely Benign | 0.2 | 0.66 | Ambiguous | 0.20 | Likely Benign | 0.01 | Likely Benign | 0.027 | Likely Benign | -0.21 | Neutral | 0.198 | Benign | 0.114 | Benign | 1.77 | Pathogenic | 0.45 | Tolerated | 3.38 | 23 | 0.0514 | 0.3207 | 3 | 4 | 0.3 | 14.03 | |||||||||||||||||||||||||
| c.1118G>T | G373V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G373V is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6‑33438023‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are FoldX, Foldetta, and SIFT, while Rosetta is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as pathogenic. Overall, the majority of predictions support a benign impact, and this consensus does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | Uncertain | 1 | 6-33438023-G-T | 6 | 5.03e-6 | -6.062 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 5.32 | Destabilizing | 3.2 | 0.82 | Ambiguous | 3.07 | Destabilizing | 0.09 | Likely Benign | 0.428 | Likely Benign | -0.98 | Neutral | 0.007 | Benign | 0.001 | Benign | 3.90 | Benign | 0.00 | Affected | 3.53 | 16 | 0.1424 | 0.4004 | -1 | -3 | 4.6 | 42.08 | 207.6 | -68.1 | 1.9 | 1.1 | -0.6 | 0.1 | Uncertain | Gly373 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are observed in the variant simulations, Val373 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effect on the Gly-rich Ω loop dynamics can only be studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1151G>C | G384A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G384A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include REVEL, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and Rosetta. Predictions from FoldX and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | -6.927 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 1.81 | Ambiguous | 0.2 | 2.16 | Destabilizing | 1.99 | Ambiguous | 0.04 | Likely Benign | 0.307 | Likely Benign | -0.40 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 1.33 | Pathogenic | 0.05 | Affected | 0.3986 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1157G>C | G386A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change G386A has no ClinVar entry and is not reported in gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (SGM‑Consensus, REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar) and pathogenic (FoldX, polyPhen‑2 HumDiv, SIFT). Two tools report uncertainty: Rosetta and Foldetta. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, while Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a benign effect for G386A. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.733139 | Disordered | 0.424156 | Uncertain | 0.334 | 0.898 | 0.750 | -6.453 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 2.14 | Destabilizing | 0.7 | 1.05 | Ambiguous | 1.60 | Ambiguous | 0.14 | Likely Benign | 0.331 | Likely Benign | -0.55 | Neutral | 0.718 | Possibly Damaging | 0.332 | Benign | 3.93 | Benign | 0.05 | Affected | 0.3815 | 0.4868 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1480A>G | I494V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I494V is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33438512‑A‑G). Functional prediction tools that agree on benign impact include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Pathogenic predictions come from premPS and FATHMM. Predictions that are inconclusive are FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign; Foldetta remains uncertain. Overall, the majority of evidence supports a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.155435 | Structured | 0.353330 | Uncertain | 0.941 | 0.157 | 0.000 | Conflicting | 2 | 6-33438512-A-G | 36 | 2.23e-5 | -7.102 | In-Between | 0.112 | Likely Benign | Likely Benign | 1.16 | Ambiguous | 0.0 | 0.71 | Ambiguous | 0.94 | Ambiguous | 1.02 | Destabilizing | 0.439 | Likely Benign | -0.83 | Neutral | 0.278 | Benign | 0.179 | Benign | -1.30 | Pathogenic | 0.07 | Tolerated | 3.37 | 35 | 0.0965 | 0.2491 | 4 | 3 | -0.3 | -14.03 | 248.6 | 29.3 | 0.0 | 0.0 | -1.1 | 0.5 | X | Potentially Benign | The sec-butyl side chain of Ile494, located in an α-helix (res. Leu489-Glu519), packs against hydrophobic residues (e.g., Phe484, Leu465, Trp572, Ala493, Met468) in an inter-helix space (res. Leu489-Glu519 and res. Ala461-Phe476). In the variant simulations, the hydrophobic iso-propyl side chain of Val494, which is of a similar size and has similar physicochemical properties to Ile494 in the WT, resides similarly in the inter-helix hydrophobic space. Thus, no negative effects on the protein structure are observed. | ||||||||||||||
| c.1614G>C | E538D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E538D is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and FATHMM. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports benign. Consequently, the variant is most likely benign, and this prediction does not contradict the ClinVar status (no ClinVar entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.122885 | Structured | 0.033501 | Uncertain | 0.938 | 0.359 | 0.000 | -2.355 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.0 | 0.09 | Likely Benign | 0.21 | Likely Benign | 0.16 | Likely Benign | 0.122 | Likely Benign | -0.88 | Neutral | 0.002 | Benign | 0.005 | Benign | 3.35 | Benign | 0.30 | Tolerated | 0.1801 | 0.2352 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1614G>T | E538D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E538D is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and FATHMM. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports benign. Consequently, the variant is most likely benign, and this prediction does not contradict the ClinVar status (no ClinVar entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.122885 | Structured | 0.033501 | Uncertain | 0.938 | 0.359 | 0.000 | -2.355 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.0 | 0.09 | Likely Benign | 0.21 | Likely Benign | 0.16 | Likely Benign | 0.122 | Likely Benign | -0.88 | Neutral | 0.002 | Benign | 0.005 | Benign | 3.35 | Benign | 0.30 | Tolerated | 0.1801 | 0.2352 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1939G>T | G647C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647C is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, the majority (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) uniformly predict a benign effect, whereas only SIFT classifies the change as pathogenic. High‑accuracy tools give consistent benign results: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign stability change. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -2.955 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.0 | -0.20 | Likely Benign | 0.11 | Likely Benign | 0.05 | Likely Benign | 0.112 | Likely Benign | -1.63 | Neutral | 0.003 | Benign | 0.004 | Benign | 3.44 | Benign | 0.02 | Affected | 0.1194 | 0.3250 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.221G>A | S74N 2D AIThe SynGAP1 missense variant S74N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33425829‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No Foldetta stability result is available. Overall, the majority of computational evidence indicates that the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | Uncertain | 1 | 6-33425829-G-A | 5 | 3.10e-6 | -5.156 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -0.89 | Neutral | 0.043 | Benign | 0.007 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1434 | 0.4193 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||
| c.2225G>C | R742P 2D  AIThe SynGAP1 missense variant R742P is catalogued in gnomAD (6-33441690‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign or likely benign outcome. Only SIFT classifies the change as pathogenic, representing the sole discordant prediction. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise reports likely benign. The Foldetta protein‑folding stability analysis is not available for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | 6-33441690-G-C | 1 | 6.20e-7 | -2.920 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -0.51 | Neutral | 0.001 | Benign | 0.004 | Benign | 2.84 | Benign | 0.03 | Affected | 4.32 | 2 | 0.2598 | 0.3637 | -2 | 0 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||
| c.2299A>C | I767L 2D AIThe SynGAP1 missense variant I767L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -1.881 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -0.73 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.13 | Benign | 0.34 | Tolerated | 0.1020 | 0.4317 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2311T>A | S771T 2D AIThe SynGAP1 missense variant S771T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -4.765 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -1.38 | Neutral | 0.649 | Possibly Damaging | 0.433 | Benign | 4.07 | Benign | 0.23 | Tolerated | 0.1497 | 0.6310 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2311T>C | S771P 2D AIThe SynGAP1 missense variant S771P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -5.045 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.180 | Likely Benign | -1.32 | Neutral | 0.901 | Possibly Damaging | 0.692 | Possibly Damaging | 4.03 | Benign | 0.19 | Tolerated | 0.2194 | 0.5515 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2506A>G | S836G 2D AIThe SynGAP1 missense variant S836G is listed in ClinVar (ID 537003.0) with an uncertain significance annotation and is observed in the gnomAD database (variant ID 6‑33443058‑A‑G). Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Benign. Foldetta, a protein‑folding stability predictor, did not return a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign classification, which does not conflict with the ClinVar uncertain designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.634582 | Binding | 0.269 | 0.859 | 0.250 | Uncertain | 1 | 6-33443058-A-G | 4 | 2.48e-6 | -4.749 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -1.65 | Neutral | 0.006 | Benign | 0.019 | Benign | 2.54 | Benign | 0.39 | Tolerated | 3.77 | 5 | 0.2567 | 0.4089 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||
| c.26A>G | H9R 2D AIThe SynGAP1 missense variant H9R is reported in gnomAD (variant ID 6‑33420290‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H9R is most likely benign, and this conclusion does not contradict any ClinVar status (none is provided). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | 6-33420290-A-G | -1.736 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.04 | Neutral | 0.012 | Benign | 0.002 | Benign | 4.25 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2330 | 0.3266 | 0 | 2 | -1.3 | 19.05 | ||||||||||||||||||||||||||||||||||||
| c.2710A>G | M904V 2D AIThe SynGAP1 missense variant M904V is reported in ClinVar (ID 833650.0) as benign and is present in gnomAD (variant ID 6‑33443262‑A‑G). All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence strongly supports a benign classification, which aligns with the ClinVar status and shows no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.589073 | Binding | 0.350 | 0.920 | 0.250 | Likely Benign | 2 | 6-33443262-A-G | 77 | 4.78e-5 | -2.907 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.33 | Neutral | 0.039 | Benign | 0.023 | Benign | 2.80 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.3724 | 0.3948 | 2 | 1 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||||
| c.2753C>T | A918V 2D AIThe SynGAP1 missense variant A918V is listed in ClinVar with an “Uncertain” status and is present in gnomAD (gnomAD ID 6‑33443305‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; a Foldetta stability prediction is not available. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.632174 | Disordered | 0.891459 | Binding | 0.317 | 0.860 | 0.250 | Uncertain | 3 | 6-33443305-C-T | 2 | 1.24e-6 | -3.684 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -1.61 | Neutral | 0.980 | Probably Damaging | 0.782 | Possibly Damaging | 2.61 | Benign | 0.03 | Affected | 4.32 | 4 | 0.1226 | 0.5586 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||
| c.2833C>T | H945Y 2D  AIThe SynGAP1 missense variant H945Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for H945Y, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.849210 | Binding | 0.386 | 0.923 | 0.750 | -6.036 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.335 | Likely Benign | -0.07 | Neutral | 0.982 | Probably Damaging | 0.903 | Possibly Damaging | 5.27 | Benign | 0.05 | Affected | 0.2102 | 0.3912 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.283C>T | H95Y 2D AIThe SynGAP1 missense variant H95Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that H95Y is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.590542 | Binding | 0.335 | 0.875 | 0.625 | -3.610 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.090 | Likely Benign | -1.45 | Neutral | 0.219 | Benign | 0.014 | Benign | 4.14 | Benign | 0.00 | Affected | 0.0880 | 0.4122 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3349G>T | G1117C 2D AIThe SynGAP1 missense variant G1117C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -9.045 | Likely Pathogenic | 0.112 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -1.30 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 4.56 | Benign | 0.03 | Affected | 0.1347 | 0.4612 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3358G>T | G1120C 2D AIThe SynGAP1 missense variant G1120C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for G1120C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | -9.324 | Likely Pathogenic | 0.112 | Likely Benign | Likely Benign | 0.311 | Likely Benign | -1.32 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 3.60 | Benign | 0.03 | Affected | 0.1271 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.367G>C | A123P 2D AIThe SynGAP1 missense variant A123P is listed in ClinVar (ID 4767034) with an “Uncertain” status and is not reported in gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, SIFT). The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a “Likely Benign” outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for A123P, and this conclusion does not contradict the current ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.689505 | Binding | 0.324 | 0.886 | 0.750 | Uncertain | 1 | -2.930 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.166 | Likely Benign | -1.14 | Neutral | 0.838 | Possibly Damaging | 0.278 | Benign | 4.14 | Benign | 0.02 | Affected | 0.2301 | 0.5999 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||
| c.3839T>C | M1280T 2D AIThe SynGAP1 missense variant M1280T is catalogued in gnomAD (6‑33447887‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign or likely benign outcome. In contrast, SIFT and FATHMM predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | 6-33447887-T-C | 3 | 1.93e-6 | -2.305 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.158 | Likely Benign | -2.26 | Neutral | 0.369 | Benign | 0.120 | Benign | 2.34 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1814 | 0.2031 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.4021G>C | A1341P 2D AIThe SynGAP1 missense variant A1341P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.980111 | Binding | 0.383 | 0.696 | 1.000 | -3.168 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.59 | Neutral | 0.131 | Benign | 0.057 | Benign | 4.02 | Benign | 0.02 | Affected | 0.2154 | 0.5703 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.709G>T | A237S 2D AIThe SynGAP1 missense variant A237S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and FATHMM. No tool predicts a pathogenic outcome; the only inconclusive results come from premPS and ESM1b, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -7.696 | In-Between | 0.112 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.6 | 0.34 | Likely Benign | 0.41 | Likely Benign | 0.61 | Ambiguous | 0.421 | Likely Benign | -1.41 | Neutral | 0.259 | Benign | 0.048 | Benign | 5.82 | Benign | 0.31 | Tolerated | 0.2209 | 0.4357 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1061C>G | A354G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A354G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. Predictions marked uncertain (Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, while Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a benign impact for A354G, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | -4.285 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.0 | 1.24 | Ambiguous | 0.83 | Ambiguous | 0.65 | Ambiguous | 0.037 | Likely Benign | -1.87 | Neutral | 0.146 | Benign | 0.038 | Benign | 1.75 | Pathogenic | 0.05 | Affected | 0.2563 | 0.4777 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.2008C>G | L670V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L670V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score all indicate benign. Only ESM1b predicts pathogenicity, while FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta remains uncertain. Taken together, the overwhelming majority of evidence supports a benign interpretation, and this conclusion does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | -9.829 | Likely Pathogenic | 0.113 | Likely Benign | Likely Benign | 1.45 | Ambiguous | 0.1 | 1.05 | Ambiguous | 1.25 | Ambiguous | 0.15 | Likely Benign | 0.025 | Likely Benign | -0.60 | Neutral | 0.127 | Benign | 0.023 | Benign | 3.67 | Benign | 0.74 | Tolerated | 0.1558 | 0.4108 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2119G>T | A707S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A707S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign. In contrast, polyPhen‑2 HumDiv and HumVar classify the variant as pathogenic. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence supports a benign impact, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | -4.432 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.70 | Ambiguous | 0.1 | 0.63 | Ambiguous | 0.67 | Ambiguous | 0.28 | Likely Benign | 0.150 | Likely Benign | -1.07 | Neutral | 0.848 | Possibly Damaging | 0.945 | Probably Damaging | 3.43 | Benign | 0.08 | Tolerated | 0.1825 | 0.2924 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.2213G>T | S738I 2D  AIThe SynGAP1 missense variant S738I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -4.312 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.78 | Neutral | 0.642 | Possibly Damaging | 0.393 | Benign | 2.66 | Benign | 0.01 | Affected | 0.0847 | 0.3636 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2237T>G | V746G 2D AIThe SynGAP1 missense variant V746G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, V746G is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -2.971 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.89 | Neutral | 0.136 | Benign | 0.270 | Benign | 2.74 | Benign | 0.02 | Affected | 0.1897 | 0.2550 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2378A>G | K793R 2D AIThe SynGAP1 missense variant K793R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess sequence conservation and functional impact all converge on a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool in the dataset indicates pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign,” while Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | -2.789 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.29 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.18 | Benign | 0.22 | Tolerated | 0.5270 | 0.1657 | Weaken | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||||
| c.2399G>T | G800V 2D AIThe SynGAP1 missense variant G800V is predicted to be benign by all evaluated in‑silico tools. ClinVar contains no entry for this variant, and it is not reported in gnomAD. Consensus predictions from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a likely benign outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a pathogenic ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.852992 | Disordered | 0.588350 | Binding | 0.303 | 0.884 | 0.625 | -4.890 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.24 | Neutral | 0.451 | Benign | 0.157 | Benign | 2.73 | Benign | 0.25 | Tolerated | 0.1130 | 0.4198 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2767A>G | I923V 2D AIThe SynGAP1 missense variant I923V is reported in gnomAD (ID 6‑33443319‑A‑G) and has no ClinVar entry. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this is consistent with the absence of a ClinVar pathogenic classification. Therefore, the variant is most likely benign, with no conflict with ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.964857 | Binding | 0.292 | 0.852 | 0.250 | 6-33443319-A-G | 1 | 6.20e-7 | -2.010 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.059 | Likely Benign | 0.05 | Neutral | 0.028 | Benign | 0.009 | Benign | 2.76 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.1589 | 0.4060 | 3 | 4 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.2851C>T | H951Y 2D AIThe SynGAP1 missense variant H951Y is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the consensus of high‑accuracy predictors (AlphaMissense‑Optimized and SGM‑Consensus) supports a benign interpretation. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.901477 | Binding | 0.415 | 0.925 | 0.750 | -5.165 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.142 | Likely Benign | -1.03 | Neutral | 0.000 | Benign | 0.001 | Benign | 5.61 | Benign | 0.10 | Tolerated | 0.2247 | 0.4112 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.293A>T | H98L 2D AIThe SynGAP1 H98L missense variant is reported in gnomAD (ID 6‑33425901‑A‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for H98L, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | 6-33425901-A-T | 1 | 6.32e-7 | -1.804 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.194 | Likely Benign | -0.51 | Neutral | 0.115 | Benign | 0.012 | Benign | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1125 | 0.6291 | -3 | -2 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||
| c.3376G>T | G1126C 2D AIThe SynGAP1 missense variant G1126C is listed in ClinVar (ID 469157.0) with an “Uncertain” status and is present in gnomAD (6‑33443928‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are SIFT and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | Uncertain | 1 | 6-33443928-G-T | 11 | 7.35e-6 | -9.389 | Likely Pathogenic | 0.113 | Likely Benign | Likely Benign | 0.449 | Likely Benign | -1.40 | Neutral | 0.005 | Benign | 0.005 | Benign | 4.74 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1326 | 0.4427 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||
| c.1088A>T | Y363F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y363F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, seven tools predict benign while four predict pathogenic, with no evidence of pathogenicity from the most accurate methods. Therefore, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -6.621 | Likely Benign | 0.114 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.42 | Likely Benign | 0.10 | Likely Benign | 0.58 | Ambiguous | 0.293 | Likely Benign | -3.42 | Deleterious | 0.997 | Probably Damaging | 0.970 | Probably Damaging | 1.74 | Pathogenic | 0.18 | Tolerated | 0.2781 | 0.3583 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.10T>C | S4P 2D AIThe SynGAP1 missense variant S4P is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools largely support a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, two tools—polyPhen‑2 HumDiv and SIFT—predict a pathogenic impact. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.131 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -0.33 | Neutral | 0.676 | Possibly Damaging | 0.307 | Benign | 4.12 | Benign | 0.00 | Affected | 0.2043 | 0.6112 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.1195G>A | A399T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A399T is listed in ClinVar (ID 1990638.0) as Benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX, Rosetta, and Foldetta, which are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the variant is most likely benign, and this conclusion aligns with its ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | Benign | 1 | -5.236 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 1.24 | Ambiguous | 0.1 | 0.91 | Ambiguous | 1.08 | Ambiguous | 0.49 | Likely Benign | 0.272 | Likely Benign | -0.40 | Neutral | 0.131 | Benign | 0.039 | Benign | 5.41 | Benign | 0.69 | Tolerated | 3.38 | 26 | 0.1335 | 0.6477 | 1 | 0 | -2.5 | 30.03 | 211.4 | -41.4 | 0.0 | 0.0 | 0.6 | 0.4 | X | Potentially Pathogenic | The methyl group of Ala399, located in an anti-parallel β sheet strand (res. Ala399-Ile411), is swapped for a hydroxyl-containing threonine. In the variant simulations, the hydroxyl group of Thr399 can form H-bonds with the backbone atoms of the residues in the membrane-facing loops (e.g., Gly382) in the C2 domain. Consequently, the ability of the Thr399 side chain to form H-bonds with the membrane-facing loops could adversely affect the dynamics and stability of the SynGAP-membrane association. However, since the effects on the dynamics of the membrane-facing loops can only be studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||||
| c.1222A>G | T408A 2D AISynGAP1 missense variant T408A is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv), and ESM1b. The high‑accuracy AlphaMissense‑Optimized score is benign, while the SGM consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Foldetta, a protein‑folding stability method, also predicts benign. Overall, the balance of evidence favors a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | Uncertain | 1 | -8.304 | Likely Pathogenic | 0.114 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.6 | -0.06 | Likely Benign | 0.16 | Likely Benign | 0.72 | Ambiguous | 0.118 | Likely Benign | -3.07 | Deleterious | 0.540 | Possibly Damaging | 0.131 | Benign | 4.16 | Benign | 0.14 | Tolerated | 0.3970 | 0.4674 | 1 | 0 | 2.5 | -30.03 | ||||||||||||||||||||||||||||
| c.2232G>C | Q744H 2D AIThe SynGAP1 missense variant Q744H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q744H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -4.359 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.83 | Neutral | 0.975 | Probably Damaging | 0.574 | Possibly Damaging | 2.68 | Benign | 0.01 | Affected | 0.1452 | 0.3443 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2232G>T | Q744H 2D AIThe SynGAP1 missense variant Q744H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q744H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -4.359 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.83 | Neutral | 0.975 | Probably Damaging | 0.574 | Possibly Damaging | 2.68 | Benign | 0.01 | Affected | 0.1452 | 0.3443 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2317A>C | M773L 2D AIThe SynGAP1 missense variant M773L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -3.458 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -0.75 | Neutral | 0.038 | Benign | 0.137 | Benign | 4.29 | Benign | 0.81 | Tolerated | 0.1517 | 0.3529 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2317A>T | M773L 2D AIThe SynGAP1 missense variant M773L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess pathogenicity uniformly predict a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the consensus of all available predictions points to a benign impact, and this is consistent with the lack of a ClinVar classification—there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -3.458 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -0.75 | Neutral | 0.038 | Benign | 0.137 | Benign | 4.29 | Benign | 0.81 | Tolerated | 0.1517 | 0.3529 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2498A>G | K833R 2D AIThe SynGAP1 missense variant K833R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that K833R is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.625797 | Binding | 0.315 | 0.863 | 0.375 | -2.158 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.83 | Neutral | 0.997 | Probably Damaging | 0.925 | Probably Damaging | 2.61 | Benign | 0.11 | Tolerated | 0.3864 | 0.0945 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||||||
| c.2500A>C | M834L 2D AIThe SynGAP1 missense variant M834L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.585406 | Disordered | 0.640801 | Binding | 0.258 | 0.863 | 0.375 | -1.926 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -0.97 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.05 | Benign | 0.00 | Affected | 0.1087 | 0.3331 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2500A>T | M834L 2D AIThe SynGAP1 missense variant M834L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.585406 | Disordered | 0.640801 | Binding | 0.258 | 0.863 | 0.375 | -1.926 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -0.97 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.05 | Benign | 0.00 | Affected | 0.1087 | 0.3331 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2635G>C | A879P 2D AIThe SynGAP1 missense variant A879P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that A879P is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.622695 | Binding | 0.277 | 0.874 | 0.250 | -4.317 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -1.16 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.61 | Benign | 0.23 | Tolerated | 0.1713 | 0.5004 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3041G>C | G1014A 2D AIThe SynGAP1 missense variant G1014A is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess sequence conservation and functional impact (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.914808 | Binding | 0.293 | 0.835 | 0.625 | -3.520 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -1.06 | Neutral | 0.025 | Benign | 0.022 | Benign | 2.78 | Benign | 0.36 | Tolerated | 0.3677 | 0.4682 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3164G>T | G1055V 2D AIThe SynGAP1 missense variant G1055V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -7.434 | In-Between | 0.114 | Likely Benign | Likely Benign | 0.399 | Likely Benign | 0.26 | Neutral | 0.818 | Possibly Damaging | 0.222 | Benign | 3.28 | Benign | 0.17 | Tolerated | 0.1399 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3173G>T | G1058V 2D AIThe SynGAP1 missense variant G1058V is reported in gnomAD (variant ID 6‑33443725‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | 6-33443725-G-T | 1 | 6.21e-7 | -6.877 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -0.12 | Neutral | 0.000 | Benign | 0.001 | Benign | 5.22 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1561 | 0.3694 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3200C>A | P1067Q 2D AIThe SynGAP1 missense variant P1067Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for P1067Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.767 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -2.39 | Neutral | 0.463 | Possibly Damaging | 0.087 | Benign | 2.84 | Benign | 0.01 | Affected | 0.1369 | 0.5415 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3890G>A | R1297K 2D  AIThe SynGAP1 missense variant R1297K is catalogued in gnomAD (ID 6‑33451764‑G‑A) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated or benign. Grouping by consensus, all listed predictors fall into the benign category, with no tool reporting a pathogenic outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign classification. Foldetta stability analysis is unavailable for this variant. Consequently, the aggregate evidence strongly supports a benign interpretation, and this assessment does not conflict with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.859585 | Disordered | 0.895222 | Binding | 0.511 | 0.817 | 0.625 | 6-33451764-G-A | 1 | 6.20e-7 | -3.780 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.074 | Likely Benign | 0.55 | Neutral | 0.000 | Benign | 0.004 | Benign | 2.80 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.4109 | 0.2801 | 2 | 3 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||||||
| c.3920C>A | P1307Q 2D AIThe SynGAP1 missense variant P1307Q is listed in ClinVar (ID 982827.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33451794‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.913511 | Binding | 0.491 | 0.901 | 0.875 | Uncertain | 1 | 6-33451794-C-A | -4.227 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.192 | Likely Benign | -0.88 | Neutral | 0.988 | Probably Damaging | 0.765 | Possibly Damaging | 2.82 | Benign | 0.03 | Affected | 3.77 | 5 | 0.1629 | 0.4464 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||
| c.474G>C | Q158H 2D AIThe SynGAP1 missense variant Q158H is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.527565 | Binding | 0.286 | 0.750 | 0.375 | -2.990 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.084 | Likely Benign | 0.02 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.19 | Benign | 0.51 | Tolerated | 0.1403 | 0.3774 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.474G>T | Q158H 2D AIThe SynGAP1 missense variant Q158H is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.527565 | Binding | 0.286 | 0.750 | 0.375 | -2.990 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.084 | Likely Benign | 0.02 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.19 | Benign | 0.51 | Tolerated | 0.1403 | 0.3774 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.1132G>A | G378S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G378S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as pathogenic, giving a mixed outcome. Because the predictions are evenly split and the high‑accuracy methods are contradictory, the variant’s functional impact is uncertain. Based on the available evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -5.331 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 6.75 | Destabilizing | 2.2 | 7.81 | Destabilizing | 7.28 | Destabilizing | 0.12 | Likely Benign | 0.565 | Likely Pathogenic | -0.63 | Neutral | 1.000 | Probably Damaging | 0.986 | Probably Damaging | 1.33 | Pathogenic | 0.08 | Tolerated | 0.3013 | 0.4724 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1186G>T | G396C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar; premPS is uncertain. The high‑accuracy consensus methods give a mixed signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of individual predictors and the SGM‑Consensus lean toward a benign interpretation, and the two high‑accuracy tools that are available also favor benign over pathogenic. Therefore, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -5.459 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 2.15 | Destabilizing | 0.7 | 2.52 | Destabilizing | 2.34 | Destabilizing | 0.59 | Ambiguous | 0.411 | Likely Benign | -3.00 | Deleterious | 0.983 | Probably Damaging | 0.533 | Possibly Damaging | 3.89 | Benign | 0.08 | Tolerated | 0.1181 | 0.4541 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1346G>C | S449T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449T is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts pathogenicity. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on these predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -6.262 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.1 | -0.46 | Likely Benign | -0.05 | Likely Benign | -0.15 | Likely Benign | 0.039 | Likely Benign | -1.48 | Neutral | 0.038 | Benign | 0.008 | Benign | 3.42 | Benign | 0.33 | Tolerated | 0.1219 | 0.5077 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2503C>G | L835V 2D AIThe SynGAP1 missense variant L835V is reported in gnomAD (variant ID 6‑33443055‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict the variant to be pathogenic. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.642742 | Binding | 0.319 | 0.863 | 0.125 | 6-33443055-C-G | 1 | 6.19e-7 | -3.439 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -0.70 | Neutral | 0.995 | Probably Damaging | 0.926 | Probably Damaging | 2.72 | Benign | 0.17 | Tolerated | 3.77 | 5 | 0.1492 | 0.2886 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.313T>G | S105A 2D AIThe SynGAP1 missense variant S105A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | -3.779 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.67 | Neutral | 0.012 | Benign | 0.002 | Benign | 4.11 | Benign | 0.00 | Affected | 0.5299 | 0.4214 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.3146C>A | P1049H 2D AIThe SynGAP1 missense variant P1049H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -5.427 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -2.06 | Neutral | 0.978 | Probably Damaging | 0.750 | Possibly Damaging | 2.76 | Benign | 0.01 | Affected | 0.1800 | 0.5070 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3662G>A | R1221Q 2D  AIThe SynGAP1 missense variant R1221Q is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database (ID 6‑33446654‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, SGM Consensus is likely benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.690604 | Disordered | 0.430363 | Uncertain | 0.906 | 0.539 | 0.375 | Conflicting | 2 | 6-33446654-G-A | 4 | 2.48e-6 | -5.491 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.46 | Neutral | 0.836 | Possibly Damaging | 0.153 | Benign | 2.56 | Benign | 0.12 | Tolerated | 3.77 | 5 | 0.2093 | 0.1736 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||||||
| c.3977C>T | P1326L 2D AIThe SynGAP1 missense variant P1326L is listed in ClinVar (ID 1004879.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as “Likely Benign,” and AlphaMissense‑Optimized also predicts benign. No Foldetta (FoldX‑MD/ Rosetta) stability result is available for this variant. Overall, the majority of evidence—including the high‑confidence SGM consensus and AlphaMissense‑Optimized prediction—supports a benign classification, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.887377 | Binding | 0.393 | 0.782 | 0.875 | Uncertain | 1 | -5.541 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.06 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 3.62 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2723 | 0.6113 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||
| c.460A>G | S154G 2D AIThe SynGAP1 missense variant S154G is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.508330 | Binding | 0.284 | 0.795 | 0.500 | -5.482 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.83 | Neutral | 0.006 | Benign | 0.008 | Benign | 4.05 | Benign | 0.11 | Tolerated | 0.2790 | 0.3944 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.511G>T | A171S 2D AIThe SynGAP1 missense variant A171S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.492272 | Uncertain | 0.358 | 0.652 | 0.375 | -0.011 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.054 | Likely Benign | 0.40 | Neutral | 0.002 | Benign | 0.001 | Benign | 4.33 | Benign | 0.91 | Tolerated | 0.2221 | 0.4383 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.596A>C | N199T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as benign or likely benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -4.123 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.2 | -0.10 | Likely Benign | 0.03 | Likely Benign | -0.04 | Likely Benign | 0.025 | Likely Benign | -0.82 | Neutral | 0.002 | Benign | 0.003 | Benign | 4.24 | Benign | 0.10 | Tolerated | 0.0862 | 0.6186 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.88C>T | H30Y 2D AIThe SynGAP1 H30Y missense variant (ClinVar ID 972248.0) is listed as “Uncertain” and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumVar and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.570702 | Disordered | 0.438063 | Uncertain | 0.373 | 0.883 | 0.250 | Uncertain | 1 | -3.047 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -1.84 | Neutral | 0.273 | Benign | 0.478 | Possibly Damaging | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1572 | 0.4856 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||
| c.112C>A | P38T 2D AIThe SynGAP1 missense variant P38T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -3.248 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -1.91 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.06 | Benign | 0.00 | Affected | 0.1993 | 0.6717 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.1528A>C | I510L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I510L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta)—all indicate a benign effect. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.025762 | Structured | 0.250630 | Uncertain | 0.945 | 0.273 | 0.000 | -2.362 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.2 | -0.05 | Likely Benign | 0.13 | Likely Benign | -1.01 | Stabilizing | 0.338 | Likely Benign | 0.69 | Neutral | 0.016 | Benign | 0.130 | Benign | -0.74 | Pathogenic | 1.00 | Tolerated | 0.0819 | 0.2461 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2122C>A | L708I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708I is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify the substitution as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and FATHMM. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -4.406 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.42 | Likely Benign | 0.0 | -0.21 | Likely Benign | 0.11 | Likely Benign | -0.05 | Likely Benign | 0.108 | Likely Benign | 0.02 | Neutral | 0.022 | Benign | 0.021 | Benign | 3.34 | Benign | 0.20 | Tolerated | 0.0749 | 0.2729 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2701G>A | A901T 2D AIThe SynGAP1 missense variant A901T is reported as “Likely Benign” in SGM‑Consensus and has no ClinVar entry or gnomAD allele. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.489838 | Uncertain | 0.306 | 0.917 | 0.375 | -4.708 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.95 | Neutral | 0.245 | Benign | 0.096 | Benign | 2.70 | Benign | 0.16 | Tolerated | 0.1587 | 0.7680 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2704G>A | A902T 2D AIThe SynGAP1 missense variant A902T is listed in ClinVar (ID 1027238.0) as benign and is observed in gnomAD (variant ID 6‑33443256‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of computational evidence supports a benign impact, consistent with the ClinVar annotation, and there is no contradiction between the predictions and the clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.517703 | Binding | 0.319 | 0.919 | 0.375 | Likely Benign | 1 | 6-33443256-G-A | 36 | 2.23e-5 | -4.966 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.11 | Neutral | 0.951 | Possibly Damaging | 0.617 | Possibly Damaging | 2.61 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1416 | 0.7053 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||
| c.2889T>A | H963Q 2D AIThe SynGAP1 missense variant H963Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only uncertain result comes from ESM1b. The high‑accuracy consensus methods also support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the evidence overwhelmingly indicates that H963Q is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.983973 | Binding | 0.325 | 0.886 | 0.750 | -7.798 | In-Between | 0.116 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -0.75 | Neutral | 0.411 | Benign | 0.132 | Benign | 4.17 | Benign | 0.29 | Tolerated | 0.2112 | 0.3979 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2889T>G | H963Q 2D AIThe SynGAP1 missense variant H963Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity; the only uncertain result comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and there is no ClinVar annotation to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.983973 | Binding | 0.325 | 0.886 | 0.750 | -7.798 | In-Between | 0.116 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -0.75 | Neutral | 0.411 | Benign | 0.132 | Benign | 4.17 | Benign | 0.29 | Tolerated | 0.2112 | 0.3979 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>T | P1046L 2D AIThe SynGAP1 missense variant P1046L is reported in gnomAD (ID 6‑33443689‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, SIFT and FATHMM predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors and the consensus analysis points to a benign classification. This conclusion is consistent with the absence of a ClinVar pathogenic report, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | 6-33443689-C-T | 1 | 6.20e-7 | -5.022 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -2.11 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.35 | Pathogenic | 0.05 | Affected | 3.77 | 5 | 0.2036 | 0.6442 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3178G>T | G1060C 2D AIThe SynGAP1 missense variant G1060C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for G1060C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | -9.630 | Likely Pathogenic | 0.116 | Likely Benign | Likely Benign | 0.363 | Likely Benign | -0.60 | Neutral | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 2.63 | Benign | 0.12 | Tolerated | 0.1340 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3289C>T | P1097S 2D AIThe SynGAP1 missense variant P1097S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | -3.748 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -1.04 | Neutral | 0.025 | Benign | 0.023 | Benign | 2.62 | Benign | 0.70 | Tolerated | 0.3332 | 0.5691 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3320A>T | Q1107L 2D AIThe SynGAP1 missense variant Q1107L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -3.785 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -3.27 | Deleterious | 0.006 | Benign | 0.004 | Benign | 2.53 | Benign | 0.01 | Affected | 0.0820 | 0.6447 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3629A>T | H1210L 2D AIThe SynGAP1 missense variant H1210L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools indicates that H1210L is most likely benign, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.599170 | Disordered | 0.587579 | Binding | 0.900 | 0.567 | 0.375 | -4.018 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -2.90 | Deleterious | 0.000 | Benign | 0.002 | Benign | 2.70 | Benign | 0.04 | Affected | 0.0673 | 0.4282 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3833C>T | P1278L 2D AIThe SynGAP1 missense variant P1278L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.806955 | Binding | 0.532 | 0.722 | 0.750 | -4.903 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -1.86 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.69 | Benign | 0.07 | Tolerated | 0.2338 | 0.4996 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3895C>A | L1299I 2D AIThe SynGAP1 missense variant L1299I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.896323 | Binding | 0.398 | 0.832 | 0.750 | -5.264 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.396 | Likely Benign | 0.71 | Neutral | 0.003 | Benign | 0.004 | Benign | 3.20 | Benign | 1.00 | Tolerated | 0.1014 | 0.3893 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3953T>C | L1318P 2D AIThe SynGAP1 missense variant L1318P is listed in gnomAD (ID 6‑33451827‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar, while pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.968271 | Binding | 0.399 | 0.865 | 0.750 | 6-33451827-T-C | -2.307 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -2.90 | Deleterious | 0.813 | Possibly Damaging | 0.212 | Benign | 3.98 | Benign | 0.00 | Affected | 3.77 | 5 | 0.3424 | 0.1411 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||
| c.1027G>A | V343I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33437932‑G‑A). Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to “Likely Benign” (3 benign vs. 1 pathogenic). High‑accuracy assessments are consistent: AlphaMissense‑Optimized is benign; the SGM‑Consensus is likely benign; and Foldetta, combining FoldX‑MD and Rosetta outputs, is benign. Overall, the collective evidence strongly supports a benign classification, and this does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | Uncertain | 2 | 6-33437932-G-A | 1 | 6.20e-7 | -6.020 | Likely Benign | 0.117 | Likely Benign | Likely Benign | -0.27 | Likely Benign | 0.0 | -0.04 | Likely Benign | -0.16 | Likely Benign | -0.39 | Likely Benign | 0.020 | Likely Benign | -0.14 | Neutral | 0.159 | Benign | 0.084 | Benign | 1.98 | Pathogenic | 0.27 | Tolerated | 3.37 | 25 | 0.1095 | 0.4536 | 4 | 3 | 0.3 | 14.03 | 240.2 | -26.9 | -0.2 | 0.2 | -0.2 | 0.2 | X | Potentially Benign | The iso-propyl side chain of Val343, located in an anti-parallel β sheet strand (res. Gly341-Pro349), is packing against multiple hydrophobic residues of the C2 domain (e.g., Leu327, Leu274, Val365). In the variant simulations, the sec-butyl side chain of Ile343 is basically able to form the same interactions as valine due to its similar hydrophobic profile. The residue swap also does not seem to cause negative effects on the protein structure based on the simulations. | |||||||||||||
| c.1055C>A | T352N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T352N is listed in ClinVar as Benign (ClinVar ID 590151.0) and is present in the gnomAD database (gnomAD ID 6‑33437960‑C‑A). Across the broad panel of in‑silico predictors, 13 tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) uniformly report a benign effect, whereas only FATHMM predicts pathogenicity. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is also benign. No predictions or stability analyses are missing or inconclusive. Overall, the computational evidence strongly supports a benign classification, consistent with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | Likely Benign | 1 | 6-33437960-C-A | 2 | 1.24e-6 | -4.817 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.20 | Likely Benign | 0.0 | -0.04 | Likely Benign | 0.08 | Likely Benign | 0.45 | Likely Benign | 0.027 | Likely Benign | -0.92 | Neutral | 0.255 | Benign | 0.057 | Benign | 1.75 | Pathogenic | 0.19 | Tolerated | 3.37 | 25 | 0.1311 | 0.4358 | 0 | 0 | -2.8 | 13.00 | 208.4 | -14.5 | -0.2 | 0.1 | -0.1 | 0.0 | X | Potentially Benign | Thr352 is located in a short α helical section within a loop connecting two β strands (res. Gly341-Pro349, res. Thr359-Pro364) originating from two different anti-parallel β sheets of the C2 domain. In the WT simulations, the side chain hydroxyl and backbone amide groups of Thr354 form hydrogen bonds with the backbone carbonyl group of Pro349 at the end of the preceding β strand. This arrangement likely stabilizes the α helical section and aids in folding, keeping the short secondary structure element intact in the variant simulations. However, the carboxamide group of the Asn352 side chain does not form hydrogen bonds with the backbone carbonyl group of Pro349. Instead, it packs against the cyclic ring and forms hydrogen bonds with the phenol group of the Tyr363 side chain in the other β strand. | |||||||||||||
| c.1177G>A | G393S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G393S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from FoldX, polyPhen‑2 HumDiv, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta) as uncertain. No other tools provide decisive evidence. Overall, the majority of reliable predictors lean toward a benign effect, and this consensus does not conflict with the absence of ClinVar annotation. Therefore, G393S is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -5.207 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 2.43 | Destabilizing | 0.7 | -0.78 | Ambiguous | 0.83 | Ambiguous | 0.24 | Likely Benign | 0.466 | Likely Benign | -1.76 | Neutral | 0.889 | Possibly Damaging | 0.444 | Benign | 1.33 | Pathogenic | 0.07 | Tolerated | 0.3011 | 0.5232 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1975T>G | S659A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S659A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while ESM1b remains uncertain. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.067594 | Structured | 0.154597 | Uncertain | 0.954 | 0.283 | 0.000 | -7.066 | In-Between | 0.117 | Likely Benign | Likely Benign | -0.31 | Likely Benign | 0.0 | 0.02 | Likely Benign | -0.15 | Likely Benign | 0.14 | Likely Benign | 0.088 | Likely Benign | -2.22 | Neutral | 0.097 | Benign | 0.114 | Benign | 3.50 | Benign | 0.64 | Tolerated | 0.4842 | 0.3813 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.2026A>G | S676G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and SIFT. Two tools (ESM1b and premPS) returned uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, and Foldetta predicts a benign stability change. No prediction or folding result is missing or inconclusive. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -7.249 | In-Between | 0.117 | Likely Benign | Likely Benign | 0.38 | Likely Benign | 0.1 | 0.25 | Likely Benign | 0.32 | Likely Benign | 0.67 | Ambiguous | 0.132 | Likely Benign | -2.74 | Deleterious | 0.855 | Possibly Damaging | 0.100 | Benign | 3.39 | Benign | 0.02 | Affected | 0.2772 | 0.4342 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||
| c.2218C>G | R740G 2D AIThe SynGAP1 missense variant R740G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | -4.556 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.138 | Likely Benign | -2.55 | Deleterious | 0.993 | Probably Damaging | 0.887 | Possibly Damaging | 2.56 | Benign | 0.03 | Affected | 0.3739 | 0.3149 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2315T>A | F772Y 2D AIThe SynGAP1 missense variant F772Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -2.657 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.54 | Neutral | 0.705 | Possibly Damaging | 0.786 | Possibly Damaging | 4.17 | Benign | 0.35 | Tolerated | 0.1058 | 0.1892 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2737A>C | T913P 2D AIThe SynGAP1 missense variant T913P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for T913P, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.763517 | Binding | 0.339 | 0.899 | 0.375 | -2.029 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.176 | Likely Benign | -1.43 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.65 | Benign | 0.26 | Tolerated | 0.2082 | 0.5786 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.27T>A | H9Q 2D AIThe SynGAP1 missense variant H9Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | -3.477 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -0.05 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.24 | Benign | 0.00 | Affected | 0.2074 | 0.4297 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.27T>G | H9Q 2D AIThe SynGAP1 missense variant H9Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | -3.477 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.05 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.24 | Benign | 0.00 | Affected | 0.2074 | 0.4297 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2864C>G | S955C 2D AIThe SynGAP1 missense variant S955C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (5 pathogenic vs. 4 benign) indicate a likely pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant has no existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | -8.675 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -1.48 | Neutral | 0.977 | Probably Damaging | 0.796 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.1741 | 0.5470 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.2874C>A | H958Q 2D AIThe SynGAP1 missense variant H958Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.976011 | Binding | 0.371 | 0.913 | 0.750 | -8.625 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.97 | Neutral | 0.925 | Possibly Damaging | 0.316 | Benign | 4.18 | Benign | 0.11 | Tolerated | 0.2281 | 0.3736 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2874C>G | H958Q 2D AIThe SynGAP1 missense variant H958Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.976011 | Binding | 0.371 | 0.913 | 0.750 | -8.625 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.97 | Neutral | 0.925 | Possibly Damaging | 0.316 | Benign | 4.18 | Benign | 0.11 | Tolerated | 0.2281 | 0.3736 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2902G>T | G968C 2D AIThe SynGAP1 missense variant G968C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -8.441 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.170 | Likely Benign | -0.99 | Neutral | 0.992 | Probably Damaging | 0.820 | Possibly Damaging | 4.12 | Benign | 0.07 | Tolerated | 0.1294 | 0.4768 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2998A>C | I1000L 2D AIThe SynGAP1 missense variant I1000L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -2.103 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -0.34 | Neutral | 0.211 | Benign | 0.108 | Benign | 2.76 | Benign | 0.70 | Tolerated | 0.0916 | 0.4034 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.299A>G | Y100C 2D AIThe SynGAP1 missense variant Y100C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -3.789 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -0.88 | Neutral | 0.994 | Probably Damaging | 0.816 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.3073 | 0.2213 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.299A>T | Y100F 2D AIThe SynGAP1 missense variant Y100F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -3.056 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -0.50 | Neutral | 0.928 | Possibly Damaging | 0.222 | Benign | 4.21 | Benign | 0.00 | Affected | 0.2784 | 0.3087 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3170G>A | S1057N 2D AIThe SynGAP1 missense variant S1057N is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | Uncertain | 1 | -6.386 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.218 | Likely Benign | -0.41 | Neutral | 0.451 | Benign | 0.129 | Benign | 5.25 | Benign | 0.28 | Tolerated | 0.2232 | 0.4605 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||
| c.3413C>G | S1138C 2D AIThe SynGAP1 missense variant S1138C is catalogued in gnomAD (ID 6‑33444448‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for S1138C. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.738250 | Binding | 0.346 | 0.869 | 1.000 | 6-33444448-C-G | 1 | 6.20e-7 | -7.850 | In-Between | 0.117 | Likely Benign | Likely Benign | 0.425 | Likely Benign | -2.48 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 5.40 | Benign | 0.04 | Affected | 4.32 | 4 | 0.1472 | 0.6396 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||
| c.35G>C | S12T 2D AIThe SynGAP1 missense variant S12T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a benign effect. AlphaMissense‑Optimized independently scores the variant as benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.490599 | Uncertain | 0.355 | 0.916 | 0.500 | -4.304 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.16 | Neutral | 0.208 | Benign | 0.024 | Benign | 4.14 | Benign | 0.00 | Affected | 0.1487 | 0.6303 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3716C>G | A1239G 2D AIThe SynGAP1 missense variant lies within a coiled‑coil domain. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is unavailable. ClinVar contains no entry for this variant, and it is not present in gnomAD. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -4.188 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -2.09 | Neutral | 0.139 | Benign | 0.088 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.1707 | 0.3142 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3860C>T | P1287L 2D AIThe SynGAP1 missense variant P1287L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33447908‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.813701 | Binding | 0.538 | 0.777 | 0.750 | Conflicting | 2 | 6-33447908-C-T | -2.800 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -1.66 | Neutral | 0.021 | Benign | 0.017 | Benign | 2.76 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1861 | 0.4727 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3929C>T | T1310M 2D  AIThe SynGAP1 missense variant T1310M is listed in ClinVar (ID 2160201.0) as Benign and is present in gnomAD (gnomAD ID 6‑33451803‑C‑T). All evaluated in‑silico predictors report a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, which aligns with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.959076 | Binding | 0.398 | 0.904 | 0.750 | Benign | 1 | 6-33451803-C-T | 17 | 1.05e-5 | -4.822 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.069 | Likely Benign | 2.19 | Neutral | 0.021 | Benign | 0.005 | Benign | 2.98 | Benign | 0.93 | Tolerated | 3.77 | 5 | 0.1144 | 0.5397 | -1 | -1 | 2.6 | 30.09 | ||||||||||||||||||||||||||||||||
| c.3952C>A | L1318M 2D AIThe SynGAP1 missense variant L1318M is listed in gnomAD (ID 6‑33451826‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.968271 | Binding | 0.399 | 0.865 | 0.750 | 6-33451826-C-A | -4.625 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.62 | Neutral | 0.939 | Possibly Damaging | 0.396 | Benign | 4.01 | Benign | 0.03 | Affected | 3.77 | 5 | 0.1083 | 0.4027 | 2 | 4 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||
| c.3976C>A | P1326T 2D AIThe SynGAP1 missense variant P1326T is reported in gnomAD (ID 6‑33451850‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority of the same four high‑accuracy tools) is benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that P1326T is most likely benign, and this assessment does not contradict any ClinVar classification because no ClinVar variant is listed. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.887377 | Binding | 0.393 | 0.782 | 0.875 | 6-33451850-C-A | -5.755 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.135 | Likely Benign | -0.26 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.71 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1955 | 0.5517 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||
| c.676T>G | S226A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S226A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.129801 | Structured | 0.334959 | Uncertain | 0.800 | 0.324 | 0.250 | -4.665 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.06 | Likely Benign | 0.0 | 0.25 | Likely Benign | 0.16 | Likely Benign | -0.02 | Likely Benign | 0.339 | Likely Benign | -1.64 | Neutral | 0.123 | Benign | 0.050 | Benign | 5.76 | Benign | 0.06 | Tolerated | 0.4812 | 0.5599 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.865A>G | M289V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M289V is reported in ClinVar as Benign (ClinVar ID 2122760.0) and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus all predict benign, while only FATHMM predicts pathogenic. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also indicates benign. No prediction or stability result is inconclusive. Overall, the computational evidence strongly supports a benign classification, consistent with the ClinVar status and showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.127496 | Structured | 0.403499 | Uncertain | 0.886 | 0.276 | 0.000 | Benign | 1 | -4.239 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 1.09 | Ambiguous | 0.1 | -0.27 | Likely Benign | 0.41 | Likely Benign | 0.24 | Likely Benign | 0.150 | Likely Benign | -0.36 | Neutral | 0.136 | Benign | 0.054 | Benign | 1.80 | Pathogenic | 1.00 | Tolerated | 3.38 | 23 | 0.2280 | 0.2808 | 2 | 1 | 2.3 | -32.06 | 204.2 | 51.0 | 0.0 | 0.0 | 0.2 | 0.0 | X | Potentially Benign | The hydrophobic residue Met289, located in a β hairpin linking two anti-parallel β sheet strands (res. Met289-Arg299, res. Arg272-Leu286), is swapped for another hydrophobic residue, valine. In the variant simulations, the branched hydrocarbon side chain of Val289 packs against the phenol group of the Tyr291 side chain but is unable to form methionine-aromatic interactions. β hairpins are potential nucleation sites during the initial stages of protein folding, so even minor changes in them could be significant. However, based on the simulations, the residue swap does not cause adverse effects on the structure. | ||||||||||||||||
| c.89A>T | H30L 2D AIThe SynGAP1 H30L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.570702 | Disordered | 0.438063 | Uncertain | 0.373 | 0.883 | 0.250 | -2.073 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.163 | Likely Benign | -2.88 | Deleterious | 0.462 | Possibly Damaging | 0.599 | Possibly Damaging | 3.94 | Benign | 0.00 | Affected | 0.1523 | 0.5793 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.1135T>C | S379P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S379P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, Rosetta, and Foldetta; FoldX is uncertain. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of evidence—including the high‑accuracy benign predictions—suggests that the variant is most likely benign. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.433206 | Uncertain | 0.327 | 0.931 | 0.625 | -5.007 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 1.10 | Ambiguous | 0.8 | 2.92 | Destabilizing | 2.01 | Destabilizing | 0.17 | Likely Benign | 0.430 | Likely Benign | -0.41 | Neutral | 0.808 | Possibly Damaging | 0.212 | Benign | 3.83 | Benign | 0.10 | Tolerated | 0.3035 | 0.6594 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.2596G>A | V866I 2D AIThe SynGAP1 missense variant V866I is listed in ClinVar with an “Uncertain” status (ClinVar ID 536995.0) and is present in gnomAD (6‑33443148‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.638070 | Binding | 0.266 | 0.788 | 0.250 | Conflicting | 3 | 6-33443148-G-A | 5 | 3.10e-6 | -4.652 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -0.39 | Neutral | 0.957 | Probably Damaging | 0.541 | Possibly Damaging | 2.69 | Benign | 0.27 | Tolerated | 3.82 | 4 | 0.0699 | 0.4004 | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||||
| c.2834A>G | H945R 2D AIThe SynGAP1 missense variant H945R is reported in gnomAD (variant ID 6‑33443386‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.849210 | Binding | 0.386 | 0.923 | 0.750 | 6-33443386-A-G | 1 | 6.20e-7 | -5.186 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.333 | Likely Benign | -0.59 | Neutral | 0.982 | Probably Damaging | 0.903 | Possibly Damaging | 5.05 | Benign | 0.08 | Tolerated | 4.32 | 4 | 0.2764 | 0.3020 | 0 | 2 | -1.3 | 19.05 | ||||||||||||||||||||||||||||||||||
| c.2883C>A | H961Q 2D AIThe SynGAP1 missense variant H961Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT and ESM1b predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict (3 benign vs. 1 pathogenic). High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.989835 | Disordered | 0.984562 | Binding | 0.323 | 0.893 | 0.750 | -8.368 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.49 | Neutral | 0.255 | Benign | 0.105 | Benign | 4.17 | Benign | 0.02 | Affected | 0.2022 | 0.3743 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2883C>G | H961Q 2D AIThe SynGAP1 missense variant H961Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT and ESM1b predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a majority‑benign vote and is reported as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the available predictions indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.989835 | Disordered | 0.984562 | Binding | 0.323 | 0.893 | 0.750 | -8.368 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.49 | Neutral | 0.255 | Benign | 0.105 | Benign | 4.17 | Benign | 0.02 | Affected | 0.2022 | 0.3743 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2929G>A | A977T 2D AIThe SynGAP1 missense variant A977T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -3.814 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -0.84 | Neutral | 0.965 | Probably Damaging | 0.782 | Possibly Damaging | 4.00 | Benign | 0.02 | Affected | 0.2155 | 0.6185 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2965T>A | S989T 2D AIThe SynGAP1 missense variant S989T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign outcome. No Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -3.995 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -1.48 | Neutral | 0.770 | Possibly Damaging | 0.396 | Benign | 2.66 | Benign | 0.00 | Affected | 0.1183 | 0.5377 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3160G>T | G1054C 2D AIThe SynGAP1 missense variant G1054C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -9.548 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.297 | Likely Benign | -0.59 | Neutral | 0.999 | Probably Damaging | 0.907 | Possibly Damaging | 4.00 | Benign | 0.10 | Tolerated | 0.1413 | 0.4427 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3299G>T | G1100V 2D AIThe SynGAP1 missense variant G1100V is reported in gnomAD (variant ID 6‑33443851‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign); pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the balance of evidence favors a benign classification for G1100V, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | 6-33443851-G-T | 1 | 6.51e-7 | -2.362 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.198 | Likely Benign | -2.43 | Neutral | 0.992 | Probably Damaging | 0.906 | Possibly Damaging | 1.93 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1297 | 0.4036 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3344T>A | I1115N 2D  AIThe SynGAP1 missense variant I1115N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | -4.018 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -0.60 | Neutral | 0.009 | Benign | 0.011 | Benign | 2.77 | Benign | 0.08 | Tolerated | 0.1299 | 0.1270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3367G>T | G1123C 2D AIThe SynGAP1 missense variant G1123C is listed in gnomAD (ID 6‑33443919‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy tools—supports a benign classification. This conclusion is not contradicted by ClinVar, which contains no record for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | 6-33443919-G-T | 1 | 6.64e-7 | -9.329 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.353 | Likely Benign | -1.17 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 4.34 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1348 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.3941C>T | P1314L 2D AIThe SynGAP1 missense variant P1314L is listed in ClinVar as a benign alteration (ClinVar ID 646689.0) and is present in the gnomAD database (gnomAD ID 6‑33451815‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, which is consistent with the ClinVar classification. Thus, the variant is most likely benign and does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.971592 | Binding | 0.467 | 0.903 | 0.750 | Likely Benign | 1 | 6-33451815-C-T | 2 | 1.24e-6 | -4.040 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -0.20 | Neutral | 0.421 | Benign | 0.066 | Benign | 4.19 | Benign | 0.05 | Affected | 3.77 | 5 | 0.2310 | 0.5967 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.457A>T | T153S 2D AIThe SynGAP1 missense variant T153S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.502105 | Binding | 0.297 | 0.818 | 0.625 | -1.890 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.50 | Neutral | 0.235 | Benign | 0.039 | Benign | 4.19 | Benign | 0.14 | Tolerated | 0.3756 | 0.2886 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.458C>G | T153S 2D AIThe SynGAP1 missense variant T153S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.502105 | Binding | 0.297 | 0.818 | 0.625 | -1.890 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.50 | Neutral | 0.235 | Benign | 0.039 | Benign | 4.19 | Benign | 0.14 | Tolerated | 0.3756 | 0.2886 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1108G>T | G370C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370C has no ClinVar entry and is not reported in gnomAD. Functional prediction tools fall into two groups: benign predictions come from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from REVEL, FoldX, Rosetta, Foldetta, and FATHMM. ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction or stability result is missing. Overall, the majority of tools predict a benign effect, and the high‑accuracy consensus also leans benign, while only one high‑accuracy method (Foldetta) suggests pathogenicity. Thus, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -7.071 | In-Between | 0.119 | Likely Benign | Likely Benign | 3.01 | Destabilizing | 2.1 | 2.03 | Destabilizing | 2.52 | Destabilizing | 0.29 | Likely Benign | 0.511 | Likely Pathogenic | -1.00 | Neutral | 0.353 | Benign | 0.010 | Benign | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1245 | 0.4412 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.116A>T | Y39F 2D AIThe SynGAP1 missense variant Y39F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -3.410 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -0.78 | Neutral | 0.458 | Possibly Damaging | 0.481 | Possibly Damaging | 4.05 | Benign | 0.00 | Affected | 0.2641 | 0.3363 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2243T>A | L748Q 2D AIThe SynGAP1 missense variant L748Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for L748Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | -3.177 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.46 | Neutral | 0.912 | Possibly Damaging | 0.611 | Possibly Damaging | 2.74 | Benign | 0.01 | Affected | 0.1235 | 0.1246 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2402G>T | G801V 2D AIThe SynGAP1 missense variant G801V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G801V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.874069 | Disordered | 0.636323 | Binding | 0.320 | 0.892 | 0.625 | -4.781 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -1.64 | Neutral | 0.611 | Possibly Damaging | 0.272 | Benign | 2.70 | Benign | 0.11 | Tolerated | 0.1041 | 0.4230 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2509G>A | V837I 2D AIThe SynGAP1 missense variant V837I is reported in ClinVar as “Not submitted” and is present in gnomAD (variant ID 6-33443061‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. When predictions are grouped by consensus, the benign group contains seven tools, while the pathogenic group contains two. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that V837I is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.626284 | Binding | 0.318 | 0.871 | 0.125 | 6-33443061-G-A | 1 | 6.19e-7 | -4.299 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -0.51 | Neutral | 0.992 | Probably Damaging | 0.989 | Probably Damaging | 2.63 | Benign | 0.13 | Tolerated | 3.77 | 5 | 0.0833 | 0.3848 | 3 | 4 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||||||
| c.2560C>G | R854G 2D AIThe SynGAP1 missense variant R854G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all classify the change as benign or likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns a benign score, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. No Foldetta stability prediction is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.488780 | Uncertain | 0.277 | 0.815 | 0.750 | -2.346 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.131 | Likely Benign | -1.92 | Neutral | 0.980 | Probably Damaging | 0.818 | Possibly Damaging | 4.08 | Benign | 0.03 | Affected | 0.3469 | 0.3764 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2605C>A | L869M 2D AIThe SynGAP1 missense variant L869M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.688653 | Binding | 0.272 | 0.839 | 0.250 | -4.294 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -0.09 | Neutral | 0.149 | Benign | 0.058 | Benign | 2.61 | Benign | 0.11 | Tolerated | 0.0710 | 0.3795 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2660C>T | P887L 2D AIThe SynGAP1 missense variant P887L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.602269 | Binding | 0.348 | 0.925 | 0.500 | -4.008 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.72 | Neutral | 0.152 | Benign | 0.070 | Benign | 2.81 | Benign | 0.18 | Tolerated | 0.1800 | 0.5164 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2686G>A | G896S 2D AIThe SynGAP1 missense variant G896S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign impact, and there is no conflict with ClinVar status (which has no entry). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.412816 | Uncertain | 0.314 | 0.923 | 0.625 | -2.712 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -0.63 | Neutral | 0.896 | Possibly Damaging | 0.334 | Benign | 2.59 | Benign | 0.41 | Tolerated | 0.2684 | 0.4911 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2978C>A | P993H 2D AIThe SynGAP1 missense variant P993H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P993H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -4.535 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.93 | Neutral | 0.938 | Possibly Damaging | 0.819 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.1799 | 0.4591 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3001C>T | L1001F 2D AIThe SynGAP1 missense variant L1001F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (derived from the majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (majority vote of the four high‑confidence tools) is benign; Foldetta data are unavailable. Overall, the preponderance of evidence supports a benign classification for L1001F, and this conclusion does not conflict with ClinVar, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -4.712 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.85 | Neutral | 0.934 | Possibly Damaging | 0.617 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 0.0703 | 0.2876 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3035C>T | P1012L 2D AIThe SynGAP1 missense variant P1012L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which contains no pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.894674 | Binding | 0.319 | 0.866 | 0.625 | -4.363 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.014 | Likely Benign | -0.90 | Neutral | 0.224 | Benign | 0.131 | Benign | 2.76 | Benign | 0.12 | Tolerated | 0.2155 | 0.6388 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3181G>T | G1061C 2D AIThe SynGAP1 missense variant G1061C is listed in ClinVar (ID 536997.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443733‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence (six benign vs. four pathogenic predictions) and the two high‑accuracy tools support a benign classification. This conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | Conflicting | 2 | 6-33443733-G-T | 6 | 3.73e-6 | -9.511 | Likely Pathogenic | 0.119 | Likely Benign | Likely Benign | 0.409 | Likely Benign | -1.46 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 3.97 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1283 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||
| c.3250C>T | P1084S 2D AIThe SynGAP1 missense variant P1084S is reported in gnomAD (variant ID 6‑33443802‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443802-C-T | 1 | 6.31e-7 | -3.987 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -2.24 | Neutral | 0.481 | Possibly Damaging | 0.157 | Benign | 4.03 | Benign | 0.03 | Affected | 3.77 | 5 | 0.3102 | 0.6215 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.335G>C | G112A 2D AIThe SynGAP1 missense variant G112A is listed in ClinVar with an uncertain significance (ClinVar ID 1425533.0) and is present in gnomAD (6‑33432200‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. Foldetta results are unavailable. Overall, the majority of computational evidence indicates that the variant is most likely benign, which is consistent with its ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | Uncertain | 1 | 6-33432200-G-C | 15 | 9.30e-6 | -2.456 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -2.34 | Neutral | 0.231 | Benign | 0.054 | Benign | 4.07 | Benign | 0.00 | Affected | 3.61 | 5 | 0.3817 | 0.4429 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||
| c.347A>T | Y116F 2D AIThe SynGAP1 missense variant Y116F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign likelihood. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.670235 | Binding | 0.381 | 0.878 | 0.625 | -2.911 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.71 | Neutral | 0.539 | Possibly Damaging | 0.042 | Benign | 4.21 | Benign | 0.33 | Tolerated | 0.2632 | 0.3069 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.34A>T | S12C 2D AIThe SynGAP1 missense variant S12C is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools largely support a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, two tools—polyPhen‑2 HumDiv and SIFT—predict a pathogenic impact. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is not in conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.490599 | Uncertain | 0.355 | 0.916 | 0.500 | -5.413 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.101 | Likely Benign | 0.00 | Neutral | 0.872 | Possibly Damaging | 0.206 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1025 | 0.6092 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.1016A>G | K339R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K339R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is benign. No predictions or stability results are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -5.773 | Likely Benign | 0.120 | Likely Benign | Likely Benign | -0.15 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.16 | Likely Benign | 0.08 | Likely Benign | 0.134 | Likely Benign | -2.05 | Neutral | 0.046 | Benign | 0.040 | Benign | 1.94 | Pathogenic | 0.48 | Tolerated | 0.4197 | 0.0976 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.104T>C | V35A 2D AIThe SynGAP1 missense variant V35A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumVar and SIFT, predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -3.552 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -0.05 | Neutral | 0.267 | Benign | 0.481 | Possibly Damaging | 4.25 | Benign | 0.00 | Affected | 0.2340 | 0.2144 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.1127G>T | G376V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G376V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, matching the majority of individual benign calls. High‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts benign, SGM Consensus predicts benign, whereas Foldetta (integrating FoldX‑MD and Rosetta) predicts pathogenic. No prediction is missing or inconclusive. Overall, the balance of evidence leans toward a benign effect; this is consistent with the lack of ClinVar annotation and gnomAD presence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -6.242 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 4.84 | Destabilizing | 0.8 | -0.81 | Ambiguous | 2.02 | Destabilizing | -0.18 | Likely Benign | 0.541 | Likely Pathogenic | -0.66 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1594 | 0.3525 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1163G>T | G388V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G388V is catalogued in gnomAD (6-33438068-G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Pathogenic predictions arise from REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized remains benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates benign, whereas Foldetta—combining FoldX‑MD and Rosetta stability outputs—labels the variant as pathogenic. Overall, the majority of tools and the Foldetta result point to a pathogenic effect. This conclusion does not conflict with ClinVar, which currently contains no classification for G388V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | 6-33438068-G-T | -6.887 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 6.58 | Destabilizing | 6.6 | 5.51 | Destabilizing | 6.05 | Destabilizing | -0.11 | Likely Benign | 0.620 | Likely Pathogenic | -0.93 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 4.32 | 3 | 0.1600 | 0.3897 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||
| c.1172G>C | G391A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G391A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, polyPhen‑2 HumDiv, and FATHMM. Predictions that are inconclusive are Rosetta and Foldetta. The high‑accuracy consensus from AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also likely benign, and Foldetta remains uncertain. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -5.712 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 2.02 | Destabilizing | 0.5 | 1.92 | Ambiguous | 1.97 | Ambiguous | 0.11 | Likely Benign | 0.442 | Likely Benign | -0.76 | Neutral | 0.633 | Possibly Damaging | 0.219 | Benign | 1.33 | Pathogenic | 0.19 | Tolerated | 0.3828 | 0.4870 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1187G>T | G396V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396V is catalogued in gnomAD (6‑33438092‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, Rosetta, Foldetta, and PROVEAN. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a pathogenic effect. Overall, the majority of predictions lean toward a benign impact, and this consensus does not contradict any ClinVar status (none reported). Thus, based on the available computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | 6-33438092-G-T | 6 | 3.72e-6 | -5.663 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 3.49 | Destabilizing | 1.7 | 5.28 | Destabilizing | 4.39 | Destabilizing | 0.34 | Likely Benign | 0.332 | Likely Benign | -2.56 | Deleterious | 0.062 | Benign | 0.014 | Benign | 3.90 | Benign | 0.24 | Tolerated | 3.41 | 15 | 0.1287 | 0.4337 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||
| c.1610C>T | A537V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A537V is listed in ClinVar as Benign (ClinVar ID 766762.0) and is present in gnomAD (ID 6‑33438853‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is benign. FoldX alone is uncertain and therefore not considered evidence. Overall, the consensus of available predictions indicates that the variant is most likely benign, in agreement with its ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.037313 | Uncertain | 0.928 | 0.362 | 0.000 | Likely Benign | 1 | 6-33438853-C-T | 7 | 4.34e-6 | -6.888 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.54 | Ambiguous | 0.0 | -0.05 | Likely Benign | 0.25 | Likely Benign | 0.41 | Likely Benign | 0.382 | Likely Benign | -1.97 | Neutral | 0.977 | Probably Damaging | 0.469 | Possibly Damaging | -1.26 | Pathogenic | 0.24 | Tolerated | 3.37 | 35 | 0.1328 | 0.4748 | 0 | 0 | 2.4 | 28.05 | 220.3 | -45.1 | 0.0 | 0.0 | -0.7 | 0.1 | X | Potentially Benign | Ala537 is located on the outer surface of an α-helix (res. Ala533-Val560). The methyl group of Ala537 is on the surface and does not form any interactions. In the variant simulations, the iso-propyl side chain of Val537 is also on the surface, similar to Ala537 in the WT, causing no negative structural effects. | |||||||||||||
| c.2168C>T | T723I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723I is listed in ClinVar as Benign (ClinVar ID 436924.0) and is observed in gnomAD (variant ID 6‑33441633‑C‑T). Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT classifies the change as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates a benign impact. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | Likely Benign | 1 | 6-33441633-C-T | 2 | 1.24e-6 | -2.591 | Likely Benign | 0.120 | Likely Benign | Likely Benign | -0.39 | Likely Benign | 0.0 | -0.20 | Likely Benign | -0.30 | Likely Benign | 0.26 | Likely Benign | 0.045 | Likely Benign | -2.09 | Neutral | 0.088 | Benign | 0.030 | Benign | 3.39 | Benign | 0.03 | Affected | 3.50 | 8 | 0.0708 | 0.5803 | 0 | -1 | 5.2 | 12.05 | 252.3 | -31.6 | 0.0 | 0.0 | -0.2 | 0.2 | X | Uncertain | The hydroxyl group of Thr723, located on the outer surface of an α-helix (res. Leu714-Arg726), continuously forms hydrogen bonds with the backbone carbonyl of Asn719 in the WT simulations, potentially lowering the stability of the α-helix. In the variant simulations, the sec-butyl side chain of Ile723 cannot form any hydrogen bonds, which, in theory, could increase the helix stability. However, because the model ends abruptly at the C-terminus, no definite conclusions can be drawn based on the simulations. | |||||||||||||
| c.2206C>T | R736C 2D  AISynGAP1 missense variant R736C is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441671‑C‑T). Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence indicates a benign effect, which does not conflict with the ClinVar uncertain designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | Conflicting | 3 | 6-33441671-C-T | 8 | 4.96e-6 | -7.113 | In-Between | 0.120 | Likely Benign | Likely Benign | 0.190 | Likely Benign | -2.06 | Neutral | 0.999 | Probably Damaging | 0.825 | Possibly Damaging | 2.48 | Pathogenic | 0.00 | Affected | 4.07 | 3 | 0.3740 | 0.1691 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.2234C>A | P745H 2D AIThe SynGAP1 missense variant P745H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -5.569 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.200 | Likely Benign | -2.84 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.53 | Benign | 0.00 | Affected | 0.1912 | 0.3403 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2623G>T | A875S 2D AIThe SynGAP1 missense variant A875S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that A875S is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.632173 | Binding | 0.273 | 0.872 | 0.250 | -2.719 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.07 | Neutral | 0.945 | Possibly Damaging | 0.767 | Possibly Damaging | 2.78 | Benign | 0.08 | Tolerated | 0.2607 | 0.6338 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3158G>A | S1053N 2D AIThe SynGAP1 missense variant S1053N is reported in gnomAD (variant ID 6‑33443710‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, while the consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus also indicates a likely benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | 6-33443710-G-A | 1 | 6.21e-7 | -6.282 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.208 | Likely Benign | -0.54 | Neutral | 0.625 | Possibly Damaging | 0.193 | Benign | 5.30 | Benign | 0.34 | Tolerated | 3.77 | 5 | 0.1953 | 0.4605 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||
| c.3202T>A | L1068M 2D AIThe SynGAP1 missense variant L1068M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -5.739 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.22 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.0930 | 0.4582 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3326T>C | L1109P 2D AIThe SynGAP1 missense variant L1109P is listed in ClinVar with an uncertain significance (ClinVar ID 1730257.0) and is not reported in gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized indicates a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, which does not contradict the ClinVar uncertain status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | Conflicting | 2 | -5.313 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -0.52 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.65 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.3159 | 0.2330 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.3421C>G | P1141A 2D AIThe SynGAP1 missense variant P1141A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding‑stability method that combines FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools therefore provide mixed evidence: AlphaMissense‑Optimized indicates benign, while the consensus and stability analyses are unavailable. Overall, the majority of individual predictors (five versus four) favor a pathogenic interpretation, and this does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.930790 | Disordered | 0.716087 | Binding | 0.364 | 0.852 | 1.000 | -3.885 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -3.67 | Deleterious | 0.856 | Possibly Damaging | 0.492 | Possibly Damaging | 1.00 | Pathogenic | 0.00 | Affected | 0.3525 | 0.5135 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3908G>C | G1303A 2D AIThe SynGAP1 missense variant G1303A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.886612 | Binding | 0.429 | 0.854 | 0.875 | -2.637 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.29 | Neutral | 0.790 | Possibly Damaging | 0.433 | Benign | 2.83 | Benign | 0.11 | Tolerated | 0.3822 | 0.4092 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3953T>A | L1318Q 2D AIThe SynGAP1 missense variant L1318Q is reported in gnomAD (variant ID 6‑33451827‑T‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.968271 | Binding | 0.399 | 0.865 | 0.750 | 6-33451827-T-A | 1 | 6.32e-7 | -3.445 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -2.06 | Neutral | 0.834 | Possibly Damaging | 0.307 | Benign | 4.04 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1410 | 0.0903 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||
| c.496G>T | A166S 2D AIThe SynGAP1 missense variant A166 S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.505037 | Binding | 0.384 | 0.658 | 0.125 | -6.008 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -0.78 | Neutral | 0.399 | Benign | 0.212 | Benign | 4.07 | Benign | 0.05 | Affected | 0.2286 | 0.4146 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.998A>G | K333R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K333R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates benign stability. No prediction or folding result is missing or inconclusive. Overall, the majority of evidence points to a benign effect for K333R, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -4.897 | Likely Benign | 0.120 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.15 | Likely Benign | -0.04 | Likely Benign | 0.33 | Likely Benign | 0.232 | Likely Benign | -2.02 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.01 | Pathogenic | 0.14 | Tolerated | 0.4294 | 0.1134 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1162G>T | G388C 2D 3DClick to see structure in 3D Viewer AISynGAP1 G388C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and PROVEAN, while pathogenic predictions arise from REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized predicts benign, Foldetta predicts pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Because the majority of evidence points to pathogenicity and there is no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | -8.088 | Likely Pathogenic | 0.121 | Likely Benign | Likely Benign | 4.01 | Destabilizing | 3.7 | 4.95 | Destabilizing | 4.48 | Destabilizing | 0.03 | Likely Benign | 0.603 | Likely Pathogenic | -0.98 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1497 | 0.4112 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.2201C>A | P734Q 2D AIThe SynGAP1 missense variant P734Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | -4.392 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -1.92 | Neutral | 0.959 | Probably Damaging | 0.569 | Possibly Damaging | 2.87 | Benign | 0.06 | Tolerated | 0.1608 | 0.3364 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2206C>G | R736G 2D AIThe SynGAP1 missense variant R736G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv and SIFT predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | -4.100 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -2.05 | Neutral | 0.653 | Possibly Damaging | 0.361 | Benign | 2.51 | Benign | 0.00 | Affected | 0.3708 | 0.2554 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2234C>T | P745L 2D AIThe SynGAP1 missense variant P745L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote) as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -6.303 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.53 | Benign | 0.01 | Affected | 0.2205 | 0.5188 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2384C>A | P795H 2D AIThe SynGAP1 missense variant P795H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic outcome. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -7.717 | In-Between | 0.121 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -0.61 | Neutral | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 4.24 | Benign | 0.05 | Affected | 0.1947 | 0.5184 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||
| c.2503C>A | L835M 2D AIThe SynGAP1 missense variant L835M is listed in ClinVar (ID 2731331) with an uncertain significance designation and is not reported in gnomAD. Consensus from multiple in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) classify the substitution as pathogenic. High‑accuracy tools further support a benign interpretation: AlphaMissense‑Optimized reports benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.642742 | Binding | 0.319 | 0.863 | 0.125 | Conflicting | 2 | -4.153 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.45 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.67 | Benign | 0.12 | Tolerated | 3.77 | 5 | 0.0724 | 0.3839 | 2 | 4 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||
| c.2573G>A | S858N 2D AIThe SynGAP1 missense variant S858N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443125‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign. In contrast, polyPhen‑2 HumVar and SIFT predict pathogenicity, but these two tools are in the minority. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.482724 | Uncertain | 0.305 | 0.833 | 0.375 | Uncertain | 1 | 6-33443125-G-A | 2 | 1.24e-6 | -4.311 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -0.67 | Neutral | 0.448 | Benign | 0.846 | Possibly Damaging | 4.13 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1527 | 0.4772 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||
| c.2639C>T | A880V 2D AIThe SynGAP1 missense variant A880V is reported in gnomAD (variant ID 6‑33443191‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the change as pathogenic, creating a single discordant prediction. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign impact for A880V, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.621441 | Binding | 0.309 | 0.874 | 0.250 | 6-33443191-C-T | 1 | 6.20e-7 | -5.440 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -0.11 | Neutral | 0.761 | Possibly Damaging | 0.399 | Benign | 2.58 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.0947 | 0.5362 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.2932C>G | P978A 2D AIThe SynGAP1 missense variant P978A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -3.994 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.39 | Neutral | 0.008 | Benign | 0.010 | Benign | 4.30 | Benign | 0.07 | Tolerated | 0.3417 | 0.5891 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>G | S1033A 2D AIThe SynGAP1 missense variant S1033A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions indicates that the variant is most likely benign, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -3.107 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.06 | Neutral | 0.220 | Benign | 0.085 | Benign | 2.81 | Benign | 1.00 | Tolerated | 0.4387 | 0.5282 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3121C>T | P1041S 2D AIThe SynGAP1 missense variant P1041S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443673‑C‑T). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | Conflicting | 2 | 6-33443673-C-T | 1 | 6.20e-7 | -4.246 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.344 | Likely Benign | -2.72 | Deleterious | 0.664 | Possibly Damaging | 0.283 | Benign | 5.48 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.3199 | 0.6120 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.368C>G | A123G 2D AIThe SynGAP1 missense variant A123G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.689505 | Binding | 0.324 | 0.886 | 0.750 | -2.799 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -1.34 | Neutral | 0.421 | Benign | 0.074 | Benign | 4.13 | Benign | 0.03 | Affected | 0.2070 | 0.5039 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3824G>A | R1275Q 2D AIThe SynGAP1 missense variant R1275Q is listed in ClinVar with an uncertain significance (ClinVar ID 1720188.0) and is present in gnomAD (6‑33447872‑G‑A). Consensus from multiple in‑silico predictors shows a split: benign calls from REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic calls come from polyPhen‑2 HumDiv and SIFT. High‑accuracy tools reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports likely benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.648219 | Disordered | 0.790317 | Binding | 0.723 | 0.697 | 0.500 | Uncertain | 1 | 6-33447872-G-A | 2 | 1.29e-6 | -4.928 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -1.72 | Neutral | 0.898 | Possibly Damaging | 0.147 | Benign | 2.59 | Benign | 0.03 | Affected | 3.77 | 5 | 0.2592 | 0.1336 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.3874C>G | L1292V 2D AIThe SynGAP1 missense variant L1292V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.882643 | Binding | 0.586 | 0.800 | 0.625 | -4.943 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -0.14 | Neutral | 0.058 | Benign | 0.038 | Benign | 2.67 | Benign | 0.07 | Tolerated | 0.1769 | 0.2605 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3895C>G | L1299V 2D AIThe SynGAP1 missense variant L1299V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.896323 | Binding | 0.398 | 0.832 | 0.750 | -4.502 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.385 | Likely Benign | -0.02 | Neutral | 0.124 | Benign | 0.025 | Benign | 2.89 | Benign | 0.09 | Tolerated | 0.1726 | 0.3544 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3976C>T | P1326S 2D AIThe SynGAP1 missense variant P1326S is listed in ClinVar with no submitted interpretation and is present in gnomAD (ID 6‑33451850‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence—including the high‑accuracy tools—supports a benign classification. This conclusion does not contradict the ClinVar status, which currently has no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.887377 | Binding | 0.393 | 0.782 | 0.875 | 6-33451850-C-T | -5.221 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -0.35 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.74 | Benign | 0.00 | Affected | 3.77 | 5 | 0.3722 | 0.4563 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||
| c.904T>C | S302P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S302P is not reported in ClinVar and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only Rosetta predicts a pathogenic outcome, while FoldX and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also supports a benign classification. Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains inconclusive. Overall, the preponderance of evidence points to a benign impact for S302P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.263489 | Uncertain | 0.616 | 0.258 | 0.375 | -2.485 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 1.19 | Ambiguous | 0.4 | 2.74 | Destabilizing | 1.97 | Ambiguous | 0.14 | Likely Benign | 0.101 | Likely Benign | -0.89 | Neutral | 0.157 | Benign | 0.153 | Benign | 4.11 | Benign | 0.20 | Tolerated | 0.2522 | 0.6243 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.916G>A | V306I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant V306I is listed in ClinVar with an uncertain significance (ClinVar ID 4708871) and is present in gnomAD (ID 6‑33437821‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, SGM‑Consensus, and Rosetta. Tools that predict a pathogenic outcome are PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign effect; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports an uncertain result and is therefore not considered evidence for or against pathogenicity. Overall, the majority of predictions support a benign classification, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.363090 | Structured | 0.315026 | Uncertain | 0.896 | 0.287 | 0.125 | Conflicting | 2 | 6-33437821-G-A | 11 | 6.82e-6 | -5.989 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 1.13 | Ambiguous | 0.2 | 0.38 | Likely Benign | 0.76 | Ambiguous | 0.19 | Likely Benign | 0.154 | Likely Benign | -0.17 | Neutral | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 1.75 | Pathogenic | 0.36 | Tolerated | 3.38 | 19 | 0.0603 | 0.3461 | 3 | 4 | 0.3 | 14.03 | ||||||||||||||||||||||
| c.965C>G | A322G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A322G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. Only FATHMM predicts a pathogenic outcome. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain or inconclusive and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Based on the preponderance of benign predictions and the lack of contradictory evidence, the variant is most likely benign; this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -5.230 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.84 | Ambiguous | 0.1 | 1.43 | Ambiguous | 1.14 | Ambiguous | 0.64 | Ambiguous | 0.054 | Likely Benign | -1.07 | Neutral | 0.139 | Benign | 0.088 | Benign | 1.94 | Pathogenic | 0.19 | Tolerated | 0.2513 | 0.4683 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.965C>T | A322V 2D AIThe SynGAP1 missense variant A322V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No predictions or stability results are missing or inconclusive. Based on the overwhelming consensus of the available tools, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -4.905 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.02 | Likely Benign | 1.0 | -0.42 | Likely Benign | -0.20 | Likely Benign | 0.19 | Likely Benign | 0.052 | Likely Benign | -1.63 | Neutral | 0.270 | Benign | 0.136 | Benign | 1.96 | Pathogenic | 0.22 | Tolerated | 0.1279 | 0.6126 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.103G>T | V35F 2D AIThe SynGAP1 missense variant V35F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for V35F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -4.114 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -0.79 | Neutral | 0.923 | Possibly Damaging | 0.865 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.0765 | 0.3421 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.1061C>T | A354V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A354V is not reported in ClinVar and is present in the gnomAD database (variant ID 6‑33437966‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The high‑accuracy consensus methods support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign outcome. Predictions from FoldX and Rosetta are inconclusive and are treated as unavailable. Overall, the majority of evidence indicates that A354V is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | 6-33437966-C-T | 1 | 6.26e-7 | -6.223 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.1 | -1.02 | Ambiguous | -0.19 | Likely Benign | 0.18 | Likely Benign | 0.027 | Likely Benign | -1.11 | Neutral | 0.146 | Benign | 0.038 | Benign | 1.81 | Pathogenic | 0.05 | Affected | 3.38 | 24 | 0.1429 | 0.6388 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||
| c.1321G>A | V441I 2D AIThe SynGAP1 missense variant V441I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, while only ESM1b predicts it as pathogenic. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates benign stability. No contradictory evidence is present. Based on the collective predictions, the variant is most likely benign, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -8.773 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | -0.24 | Likely Benign | 0.3 | 0.11 | Likely Benign | -0.07 | Likely Benign | 0.25 | Likely Benign | 0.135 | Likely Benign | -0.82 | Neutral | 0.287 | Benign | 0.038 | Benign | 3.41 | Benign | 0.16 | Tolerated | 0.0694 | 0.3412 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.1598C>G | A533G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A533G resides in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome; Rosetta is uncertain and therefore not counted as evidence. High‑accuracy assessments—all of which are available—show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta as Benign, reinforcing the benign consensus. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.179055 | Structured | 0.026324 | Uncertain | 0.843 | 0.393 | 0.000 | -3.412 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.1 | 0.64 | Ambiguous | 0.48 | Likely Benign | 0.33 | Likely Benign | 0.185 | Likely Benign | -1.94 | Neutral | 0.258 | Benign | 0.099 | Benign | -1.23 | Pathogenic | 0.23 | Tolerated | 0.2348 | 0.4737 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.23T>G | I8S 2D  AIThe SynGAP1 missense variant I8S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign prediction: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -1.577 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.266 | Likely Benign | 0.18 | Neutral | 0.107 | Benign | 0.006 | Benign | 4.02 | Benign | 0.00 | Affected | 0.2726 | 0.0910 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2689T>G | S897A 2D AIThe SynGAP1 missense variant S897A is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors—including REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.418474 | Uncertain | 0.292 | 0.928 | 0.500 | -3.959 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.48 | Neutral | 0.288 | Benign | 0.208 | Benign | 2.71 | Benign | 0.81 | Tolerated | 0.4539 | 0.5684 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2968T>C | S990P 2D AIThe SynGAP1 missense variant S990P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of any ClinVar pathogenic annotation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -3.150 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.95 | Neutral | 0.586 | Possibly Damaging | 0.377 | Benign | 2.74 | Benign | 0.01 | Affected | 0.1910 | 0.5345 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3151G>T | G1051C 2D AIThe SynGAP1 missense variant G1051C is listed in ClinVar as Pathogenic and is not reported in gnomAD. Functional prediction tools show a split assessment: benign calls come from REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. High‑accuracy methods give a benign result from AlphaMissense‑Optimized; the SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is tied (2 benign vs. 2 pathogenic) and therefore inconclusive, and Foldetta’s stability prediction is unavailable. Overall, the majority of predictions lean toward a benign effect, which contradicts the ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | Likely Pathogenic | 1 | -9.050 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | 0.497 | Likely Benign | -0.90 | Neutral | 0.971 | Probably Damaging | 0.750 | Possibly Damaging | -0.74 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.1322 | 0.4612 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||
| c.3166G>T | G1056C 2D AIThe SynGAP1 missense variant G1056C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -9.974 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | 0.432 | Likely Benign | -0.70 | Neutral | 0.994 | Probably Damaging | 0.777 | Possibly Damaging | 1.83 | Pathogenic | 0.06 | Tolerated | 0.1414 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3404A>G | K1135R 2D AIThe SynGAP1 missense variant K1135R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign impact for K1135R, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.790969 | Binding | 0.303 | 0.889 | 0.875 | -2.286 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.209 | Likely Benign | -0.82 | Neutral | 0.586 | Possibly Damaging | 0.321 | Benign | 5.44 | Benign | 0.15 | Tolerated | 0.5095 | 0.1674 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.352A>G | M118V 2D AIThe SynGAP1 missense variant M118V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The prediction is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.694846 | Disordered | 0.676867 | Binding | 0.330 | 0.883 | 0.500 | -2.322 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.158 | Likely Benign | -1.23 | Neutral | 0.012 | Benign | 0.011 | Benign | 3.98 | Benign | 0.02 | Affected | 0.3326 | 0.3630 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.3692G>C | S1231T 2D AIThe SynGAP1 missense variant S1231T is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S1231T is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.490133 | Structured | 0.519419 | Binding | 0.876 | 0.544 | 0.250 | -4.166 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -1.29 | Neutral | 0.625 | Possibly Damaging | 0.252 | Benign | 2.67 | Benign | 0.31 | Tolerated | 0.1115 | 0.4579 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.86T>C | M29T 2D AIThe SynGAP1 missense variant M29T is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that M29T is most likely benign, and this conclusion does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438540 | Uncertain | 0.341 | 0.883 | 0.250 | Uncertain | 1 | -2.167 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.199 | Likely Benign | -0.37 | Neutral | 0.018 | Benign | 0.184 | Benign | 4.33 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2633 | 0.2716 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||
| c.1153T>C | S385P 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant S385P is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6-33438058‑T‑C). Prediction tools that classify the variant as benign include REVEL, Foldetta, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Predictions from FoldX and Rosetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of computational evidence supports a benign effect, which is consistent with the ClinVar uncertain status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.733139 | Disordered | 0.425480 | Uncertain | 0.341 | 0.925 | 0.750 | Uncertain | 1 | 6-33438058-T-C | -5.431 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.91 | Ambiguous | 0.6 | -0.90 | Ambiguous | 0.01 | Likely Benign | 0.19 | Likely Benign | 0.385 | Likely Benign | -0.26 | Neutral | 0.676 | Possibly Damaging | 0.693 | Possibly Damaging | 4.63 | Benign | 0.04 | Affected | 4.32 | 3 | 0.2925 | 0.6805 | 1 | -1 | -0.8 | 10.04 | 210.3 | 18.5 | 1.8 | 0.9 | 0.3 | 0.0 | Uncertain | Ser385 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and so they are rich in glycine residues, prolines, and, to a lesser extent, small hydrophilic residues to ensure maximum flexibility. Thus, the variant’s Pro385 is potentially tolerated in the Ω loop. However, since the effects on Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||||
| c.116A>G | Y39C 2D AIThe SynGAP1 missense variant Y39C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -3.109 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.203 | Likely Benign | -2.00 | Neutral | 0.943 | Possibly Damaging | 0.941 | Probably Damaging | 3.96 | Benign | 0.00 | Affected | 0.3007 | 0.2849 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.1171G>T | G391C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and PROVEAN, whereas the remaining tools—REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—predict it to be pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a pathogenic effect. This prediction does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -8.596 | Likely Pathogenic | 0.123 | Likely Benign | Likely Benign | 2.65 | Destabilizing | 0.7 | 5.03 | Destabilizing | 3.84 | Destabilizing | 0.11 | Likely Benign | 0.640 | Likely Pathogenic | -1.29 | Neutral | 1.000 | Probably Damaging | 0.970 | Probably Damaging | 1.32 | Pathogenic | 0.03 | Affected | 0.1483 | 0.4225 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1225A>G | M409V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409V is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors indicates a benign effect: REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports likely benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote) is likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, reports benign. No conflicting evidence is present. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -4.109 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 1.13 | Ambiguous | 0.2 | -0.25 | Likely Benign | 0.44 | Likely Benign | 0.37 | Likely Benign | 0.163 | Likely Benign | -0.76 | Neutral | 0.996 | Probably Damaging | 0.974 | Probably Damaging | 4.26 | Benign | 1.00 | Tolerated | 0.2706 | 0.4286 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||
| c.1229G>C | S410T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410T has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, FoldX, Foldetta, SGM‑Consensus, REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, and premPS. No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is treated as unavailable. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. **Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -6.559 | Likely Benign | 0.123 | Likely Benign | Likely Benign | -0.32 | Likely Benign | 0.1 | -0.52 | Ambiguous | -0.42 | Likely Benign | 0.00 | Likely Benign | 0.066 | Likely Benign | -0.78 | Neutral | 0.080 | Benign | 0.026 | Benign | 4.24 | Benign | 0.84 | Tolerated | 0.1371 | 0.7159 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.2071A>T | T691S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the available predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | -9.274 | Likely Pathogenic | 0.123 | Likely Benign | Likely Benign | 0.44 | Likely Benign | 0.1 | 0.23 | Likely Benign | 0.34 | Likely Benign | 0.88 | Ambiguous | 0.041 | Likely Benign | -1.67 | Neutral | 0.860 | Possibly Damaging | 0.584 | Possibly Damaging | 3.49 | Benign | 0.61 | Tolerated | 0.2213 | 0.3343 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2273A>G | Y758C 2D AIThe SynGAP1 missense variant Y758C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -5.256 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -1.91 | Neutral | 0.998 | Probably Damaging | 0.921 | Probably Damaging | 2.71 | Benign | 0.03 | Affected | 0.3375 | 0.1922 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.2368A>T | T790S 2D AIThe SynGAP1 missense variant T790S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for T790S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -3.914 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.39 | Pathogenic | 0.33 | Tolerated | 3.64 | 6 | 0.3416 | 0.4449 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||||||||
| c.2369C>G | T790S 2D AIThe SynGAP1 missense variant T790S (ClinVar ID 1020340) is listed as Uncertain in ClinVar and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus, which is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | Uncertain | 2 | -3.914 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.39 | Pathogenic | 0.33 | Tolerated | 3.64 | 6 | 0.3416 | 0.4449 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.2578G>C | V860L 2D AIThe SynGAP1 missense variant V860L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.518121 | Binding | 0.269 | 0.803 | 0.250 | -3.053 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -0.89 | Neutral | 0.124 | Benign | 0.037 | Benign | 4.17 | Benign | 0.00 | Affected | 0.1052 | 0.5446 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2648T>A | L883Q 2D AIThe SynGAP1 missense variant L883Q is reported in gnomAD (6‑33443200‑T‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is therefore most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.641952 | Binding | 0.334 | 0.886 | 0.250 | 6-33443200-T-A | 3 | 1.86e-6 | -3.559 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -0.51 | Neutral | 0.934 | Possibly Damaging | 0.637 | Possibly Damaging | 2.66 | Benign | 0.11 | Tolerated | 4.32 | 4 | 0.1119 | 0.1505 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||
| c.2746G>T | V916F 2D AIThe SynGAP1 missense variant V916F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -4.011 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -1.18 | Neutral | 0.642 | Possibly Damaging | 0.265 | Benign | 2.67 | Benign | 0.02 | Affected | 0.0729 | 0.4593 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.301C>T | H101Y 2D AIThe SynGAP1 missense variant H101Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for H101Y, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.688356 | Binding | 0.370 | 0.884 | 0.625 | -3.404 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.95 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.0862 | 0.4004 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.40C>T | P14S 2D AIThe SynGAP1 missense variant P14S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.433034 | Structured | 0.471596 | Uncertain | 0.399 | 0.909 | 0.375 | -2.352 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.02 | Neutral | 0.212 | Benign | 0.014 | Benign | 4.23 | Benign | 0.00 | Affected | 0.3935 | 0.5938 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.752A>G | K251R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K251R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Rosetta and Foldetta give uncertain results. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -4.355 | Likely Benign | 0.123 | Likely Benign | Likely Benign | -0.37 | Likely Benign | 0.0 | -0.64 | Ambiguous | -0.51 | Ambiguous | 0.14 | Likely Benign | 0.531 | Likely Pathogenic | -0.61 | Neutral | 0.970 | Probably Damaging | 0.681 | Possibly Damaging | 5.92 | Benign | 0.34 | Tolerated | 0.4756 | 0.1049 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1052C>T | A351V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A351V is not reported in ClinVar and is absent from gnomAD. Computational predictions cluster into two groups: benign (REVEL, FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen2_HumVar) and pathogenic (PROVEAN, polyPhen2_HumDiv, SIFT, ESM1b, FATHMM, SGM‑Consensus). High‑accuracy tools give a mixed signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the balance of evidence leans toward a benign effect, but the presence of several pathogenic predictions introduces uncertainty. The variant is most likely benign based on the current computational data, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -9.002 | Likely Pathogenic | 0.124 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.0 | 0.19 | Likely Benign | 0.14 | Likely Benign | 0.29 | Likely Benign | 0.052 | Likely Benign | -2.84 | Deleterious | 0.915 | Possibly Damaging | 0.321 | Benign | 1.66 | Pathogenic | 0.03 | Affected | 0.1140 | 0.6565 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.11C>G | S4C 2D AIThe SynGAP1 missense variant S4C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for S4C, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -5.210 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.106 | Likely Benign | 0.41 | Neutral | 0.880 | Possibly Damaging | 0.700 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.0976 | 0.6129 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.143T>A | F48Y 2D AIThe SynGAP1 missense variant F48Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that F48Y is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.298791 | Structured | 0.440452 | Uncertain | 0.558 | 0.707 | 0.125 | -1.983 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.20 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.26 | Benign | 0.00 | Affected | 0.1657 | 0.2778 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2249G>C | G750A 2D AIThe SynGAP1 missense variant G750A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -3.263 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -1.65 | Neutral | 0.996 | Probably Damaging | 0.887 | Possibly Damaging | 2.52 | Benign | 0.04 | Affected | 0.3683 | 0.4792 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2880C>A | H960Q 2D AIThe SynGAP1 missense variant H960Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987911 | Disordered | 0.983385 | Binding | 0.380 | 0.901 | 0.750 | -8.551 | Likely Pathogenic | 0.124 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.79 | Neutral | 0.748 | Possibly Damaging | 0.170 | Benign | 4.20 | Benign | 0.21 | Tolerated | 0.2045 | 0.3993 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2880C>G | H960Q 2D AIThe SynGAP1 missense variant H960Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987911 | Disordered | 0.983385 | Binding | 0.380 | 0.901 | 0.750 | -8.551 | Likely Pathogenic | 0.124 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.79 | Neutral | 0.748 | Possibly Damaging | 0.170 | Benign | 4.20 | Benign | 0.21 | Tolerated | 0.2045 | 0.3993 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.28C>G | R10G 2D AIThe SynGAP1 missense variant R10G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.513657 | Binding | 0.330 | 0.915 | 0.625 | -3.931 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.175 | Likely Benign | 0.48 | Neutral | 0.058 | Benign | 0.009 | Benign | 4.15 | Benign | 0.00 | Affected | 0.3796 | 0.4015 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2980A>C | K994Q 2D AIThe SynGAP1 missense variant K994Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -1.947 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -0.45 | Neutral | 0.002 | Benign | 0.004 | Benign | 4.16 | Benign | 0.03 | Affected | 0.4975 | 0.1889 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.303C>A | H101Q 2D AIThe SynGAP1 missense variant H101Q is listed in ClinVar with an uncertain significance (ClinVar ID 1307533.0) and is present in gnomAD (ID 6‑33432168‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.688356 | Binding | 0.370 | 0.884 | 0.625 | Uncertain | 1 | 6-33432168-C-A | 1 | 6.20e-7 | -2.827 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.147 | Likely Benign | -0.37 | Neutral | 0.824 | Possibly Damaging | 0.880 | Possibly Damaging | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1487 | 0.3689 | 3 | 0 | -0.3 | -9.01 | ||||||||||||||||||||||||||||||||
| c.303C>G | H101Q 2D AIThe SynGAP1 missense variant H101Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for H101Q, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.688356 | Binding | 0.370 | 0.884 | 0.625 | -2.827 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.149 | Likely Benign | -0.37 | Neutral | 0.824 | Possibly Damaging | 0.880 | Possibly Damaging | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1487 | 0.3689 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||
| c.3086A>C | Q1029P 2D AIThe SynGAP1 missense variant Q1029P is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available for this variant. Overall, the consensus of all available predictions strongly supports a benign impact, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -2.940 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -1.23 | Neutral | 0.005 | Benign | 0.015 | Benign | 2.68 | Benign | 0.15 | Tolerated | 0.1966 | 0.4986 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3409C>T | H1137Y 2D AIThe SynGAP1 missense variant H1137Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for H1137Y, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.756488 | Binding | 0.314 | 0.879 | 0.875 | -4.506 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.310 | Likely Benign | -1.93 | Neutral | 0.925 | Possibly Damaging | 0.629 | Possibly Damaging | 5.28 | Benign | 0.00 | Affected | 0.1073 | 0.5123 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3451T>A | S1151T 2D AIThe SynGAP1 missense variant S1151T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.805072 | Binding | 0.394 | 0.839 | 0.625 | -3.874 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -0.06 | Neutral | 0.798 | Possibly Damaging | 0.535 | Possibly Damaging | 2.72 | Benign | 0.30 | Tolerated | 0.1450 | 0.5903 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.4A>G | S2G 2D AIThe SynGAP1 missense variant S2G is reported in gnomAD (ID 6‑33420268‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect for this variant, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.543646 | Binding | 0.382 | 0.922 | 0.750 | 6-33420268-A-G | 1 | 6.58e-7 | -4.273 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.48 | Neutral | 0.012 | Benign | 0.002 | Benign | 4.10 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2872 | 0.5495 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||
| c.503A>C | H168P 2D AIThe SynGAP1 missense variant H168P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome, while ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Taken together, the preponderance of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.433034 | Structured | 0.502450 | Binding | 0.402 | 0.678 | 0.125 | -7.686 | In-Between | 0.124 | Likely Benign | Likely Benign | 0.279 | Likely Benign | -1.66 | Neutral | 0.072 | Benign | 0.043 | Benign | 4.19 | Benign | 0.02 | Affected | 0.2004 | 0.3960 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.109T>G | S37A 2D AIThe SynGAP1 missense variant S37A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -4.052 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.86 | Neutral | 0.140 | Benign | 0.355 | Benign | 3.98 | Benign | 0.00 | Affected | 0.5089 | 0.4970 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.1120T>C | S374P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S374P is reported in gnomAD (6‑33438025‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and Rosetta; FoldX and Foldetta are inconclusive. The high‑accuracy consensus (SGM‑Consensus) is “Likely Benign,” derived from the unanimous benign calls of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. AlphaMissense‑Optimized also predicts benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. There is no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.428948 | Uncertain | 0.333 | 0.812 | 0.625 | 6-33438025-T-C | 1 | 7.85e-7 | -4.849 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.66 | Ambiguous | 0.6 | 2.22 | Destabilizing | 1.44 | Ambiguous | 0.34 | Likely Benign | 0.388 | Likely Benign | -0.89 | Neutral | 0.396 | Benign | 0.099 | Benign | 5.30 | Benign | 0.02 | Affected | 4.32 | 13 | 0.3012 | 0.6813 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||
| c.1126G>T | G376C 2D AISynGAP1 missense variant G376C is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from Rosetta, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools report uncertainty: Foldetta and ESM1b. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign verdict; Foldetta remains uncertain. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the most accurate methods favor a benign effect. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | Uncertain | 1 | -7.686 | In-Between | 0.125 | Likely Benign | Likely Benign | 2.56 | Destabilizing | 0.5 | 0.22 | Likely Benign | 1.39 | Ambiguous | 0.16 | Likely Benign | 0.560 | Likely Pathogenic | -1.15 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1476 | 0.3929 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||
| c.1184G>T | G395V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G395V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only FoldX and SIFT predict pathogenic. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is uncertain. Taken together, the preponderance of evidence points to a benign impact for G395V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -5.617 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 2.83 | Destabilizing | 0.7 | -0.37 | Likely Benign | 1.23 | Ambiguous | 0.08 | Likely Benign | 0.288 | Likely Benign | -1.49 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.21 | Benign | 0.03 | Affected | 0.1290 | 0.4183 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.161A>G | N54S 2D AIThe SynGAP1 missense variant N54S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.196879 | Structured | 0.464669 | Uncertain | 0.504 | 0.659 | 0.000 | -5.358 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -0.17 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 4.31 | Benign | 0.00 | Affected | 0.3740 | 0.7048 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.2008C>A | L670M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L670M occurs in the GAP domain. ClinVar contains no entry for this variant, but it is present in gnomAD (ID 6‑33441267‑C‑A). Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, FATHMM, Foldetta, and the SGM‑Consensus score (Likely Benign). Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; Rosetta gives an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | 6-33441267-C-A | 2 | 1.24e-6 | -9.438 | Likely Pathogenic | 0.125 | Likely Benign | Likely Benign | -0.11 | Likely Benign | 0.0 | 0.70 | Ambiguous | 0.30 | Likely Benign | 0.06 | Likely Benign | 0.038 | Likely Benign | -0.16 | Neutral | 0.970 | Probably Damaging | 0.777 | Possibly Damaging | 3.40 | Benign | 0.25 | Tolerated | 3.39 | 27 | 0.0826 | 0.3849 | 2 | 4 | -1.9 | 18.03 | ||||||||||||||||||||||||
| c.20C>G | S7C 2D AIThe SynGAP1 missense variant S7C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, all of which are benign, and therefore SGM‑Consensus also predicts benign. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -5.066 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.111 | Likely Benign | 0.41 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1231 | 0.5672 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2401G>T | G801C 2D AIThe SynGAP1 missense variant G801C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a benign outcome; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.874069 | Disordered | 0.636323 | Binding | 0.320 | 0.892 | 0.625 | -7.542 | In-Between | 0.125 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -1.83 | Neutral | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 2.67 | Benign | 0.06 | Tolerated | 0.1039 | 0.4021 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||
| c.2621A>C | Q874P 2D AIThe SynGAP1 missense variant Q874P is reported in gnomAD (ID 6‑33443173‑A‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar classification (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.635258 | Binding | 0.289 | 0.873 | 0.250 | 6-33443173-A-C | 9 | 5.58e-6 | -4.622 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.219 | Likely Benign | -2.89 | Deleterious | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2373 | 0.5911 | -1 | 0 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||
| c.2852A>G | H951R 2D AIThe SynGAP1 missense variant H951R is listed in ClinVar as Pathogenic (ClinVar ID 1003739.0) and is not reported in gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Based on the collective predictions, the variant is most likely benign, which contradicts its ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.901477 | Binding | 0.415 | 0.925 | 0.750 | Likely Pathogenic | 1 | -4.964 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.185 | Likely Benign | -1.08 | Neutral | 0.048 | Benign | 0.029 | Benign | 5.46 | Benign | 0.24 | Tolerated | 3.77 | 5 | 0.2808 | 0.3220 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||
| c.3417G>C | Q1139H 2D AIThe SynGAP1 missense variant Q1139H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.721191 | Binding | 0.313 | 0.866 | 1.000 | -3.962 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.265 | Likely Benign | -2.40 | Neutral | 0.002 | Benign | 0.007 | Benign | 5.41 | Benign | 0.00 | Affected | 0.1313 | 0.3801 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3417G>T | Q1139H 2D AIThe SynGAP1 missense variant Q1139H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of benign predictions and the lack of contradictory evidence, the variant is most likely benign. This assessment does not conflict with ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.721191 | Binding | 0.313 | 0.866 | 1.000 | -3.962 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.265 | Likely Benign | -2.40 | Neutral | 0.002 | Benign | 0.007 | Benign | 5.41 | Benign | 0.00 | Affected | 0.1313 | 0.3801 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3770C>G | S1257C 2D AIThe SynGAP1 missense variant S1257C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus score indicates a likely benign outcome. Only SIFT predicts a pathogenic effect, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a likely benign verdict. Foldetta data are not available for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.482380 | Uncertain | 0.889 | 0.572 | 0.375 | -7.741 | In-Between | 0.125 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -2.15 | Neutral | 0.028 | Benign | 0.027 | Benign | 2.54 | Benign | 0.05 | Affected | 0.0761 | 0.4780 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||
| c.3907G>A | G1303S 2D AIThe SynGAP1 missense variant G1303S is listed in ClinVar (ID 1736068.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only polyPhen‑2 HumDiv predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.886612 | Binding | 0.429 | 0.854 | 0.875 | Uncertain | 1 | -2.271 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.155 | Likely Benign | -0.19 | Neutral | 0.649 | Possibly Damaging | 0.433 | Benign | 2.84 | Benign | 0.18 | Tolerated | 0.2605 | 0.4197 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||
| c.43G>A | A15T 2D AIThe SynGAP1 missense variant A15T is listed in ClinVar (ID 1925632.0) with an “Uncertain” clinical significance and is present in gnomAD (6‑33420307‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.466055 | Uncertain | 0.330 | 0.912 | 0.375 | Uncertain | 1 | 6-33420307-G-A | 4 | 2.60e-6 | -3.720 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -0.08 | Neutral | 0.602 | Possibly Damaging | 0.017 | Benign | 4.16 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2053 | 0.7423 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||
| c.1115G>T | G372V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G372V is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (variant ID 6-33438020-G-T). Functional prediction tools that agree on a benign effect include premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are REVEL, FoldX, Rosetta, FATHMM, and Foldetta. The high‑accuracy consensus from AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, while Foldetta (combining FoldX‑MD and Rosetta stability outputs) indicates a pathogenic effect. Overall, the majority of predictions support a benign classification, and there is no conflict with ClinVar status because the variant is not yet reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.433034 | Structured | 0.430335 | Uncertain | 0.322 | 0.774 | 0.375 | 6-33438020-G-T | -5.898 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 2.81 | Destabilizing | 0.3 | 2.91 | Destabilizing | 2.86 | Destabilizing | 0.02 | Likely Benign | 0.535 | Likely Pathogenic | -0.92 | Neutral | 0.003 | Benign | 0.000 | Benign | -0.74 | Pathogenic | 0.06 | Tolerated | 3.52 | 18 | 0.1663 | 0.4198 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||
| c.1610C>G | A537G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A537G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict pathogenic. Uncertain results come from Rosetta, Foldetta, and premPS. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized reports benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta’s stability prediction is inconclusive. Overall, the majority of evidence supports a benign impact for A537G, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.037313 | Uncertain | 0.928 | 0.362 | 0.000 | -4.302 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.40 | Likely Benign | 0.0 | 0.88 | Ambiguous | 0.64 | Ambiguous | 0.68 | Ambiguous | 0.290 | Likely Benign | -1.85 | Neutral | 0.977 | Probably Damaging | 0.672 | Possibly Damaging | -1.30 | Pathogenic | 0.36 | Tolerated | 0.2289 | 0.3387 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1771G>T | A591S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A591S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect, and the Foldetta stability analysis is inconclusive (unavailable). Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.018787 | Structured | 0.093848 | Uncertain | 0.882 | 0.185 | 0.000 | -7.535 | In-Between | 0.126 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.1 | 1.21 | Ambiguous | 0.90 | Ambiguous | 0.49 | Likely Benign | 0.083 | Likely Benign | -2.11 | Neutral | 0.034 | Benign | 0.082 | Benign | 3.52 | Benign | 0.19 | Tolerated | 0.2405 | 0.3304 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1940G>T | G647V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess sequence conservation and structural impact uniformly classify the substitution as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a tolerated change. No tool predicts pathogenicity; only FoldX and Rosetta report uncertain stability effects, which are treated as unavailable evidence. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign effect. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -4.713 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.1 | -0.59 | Ambiguous | 0.13 | Likely Benign | -0.13 | Likely Benign | 0.115 | Likely Benign | -1.77 | Neutral | 0.178 | Benign | 0.073 | Benign | 3.52 | Benign | 0.12 | Tolerated | 0.1318 | 0.3946 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2114A>G | K705R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K705R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Thus, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.134866 | Structured | 0.379324 | Uncertain | 0.922 | 0.364 | 0.000 | -4.679 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.23 | Likely Benign | 0.18 | Likely Benign | 0.094 | Likely Benign | -1.66 | Neutral | 0.960 | Probably Damaging | 0.545 | Possibly Damaging | 3.33 | Benign | 0.06 | Tolerated | 0.3456 | 0.0789 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.2219G>T | R740L 2D AIThe SynGAP1 missense variant R740L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | -4.958 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -2.30 | Neutral | 0.064 | Benign | 0.040 | Benign | 2.57 | Benign | 0.03 | Affected | 0.2341 | 0.4243 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2317A>G | M773V 2D AIThe SynGAP1 missense variant M773V has no ClinVar entry and is not reported in gnomAD. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the computational evidence indicates that M773V is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -4.353 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.234 | Likely Benign | -0.70 | Neutral | 0.038 | Benign | 0.284 | Benign | 4.30 | Benign | 0.87 | Tolerated | 0.3145 | 0.3081 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2405G>T | G802V 2D AIThe SynGAP1 missense variant G802V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign. No Foldetta stability analysis is available for this variant. Overall, the consensus of the available predictions indicates that G802V is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.894241 | Disordered | 0.681966 | Binding | 0.294 | 0.898 | 0.625 | -3.871 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.49 | Neutral | 0.411 | Benign | 0.321 | Benign | 2.67 | Benign | 0.00 | Affected | 0.1345 | 0.3533 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2591C>T | A864V 2D AIThe SynGAP1 missense variant A864V is listed in ClinVar with an uncertain significance (ClinVar ID 655662.0) and is observed in gnomAD (ID 6‑33443143‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and FATHMM. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, which does not contradict its current ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.549308 | Disordered | 0.611966 | Binding | 0.269 | 0.788 | 0.250 | Uncertain | 2 | 6-33443143-C-T | 6 | 3.72e-6 | -4.749 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -1.35 | Neutral | 0.767 | Possibly Damaging | 0.119 | Benign | 2.45 | Pathogenic | 0.30 | Tolerated | 3.82 | 4 | 0.1170 | 0.6838 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||
| c.2671C>T | L891F 2D AIThe SynGAP1 missense variant L891F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available. Overall, the majority of evidence points to a benign effect for L891F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.505861 | Binding | 0.305 | 0.923 | 0.750 | -5.650 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -1.80 | Neutral | 0.970 | Probably Damaging | 0.744 | Possibly Damaging | 2.66 | Benign | 0.04 | Affected | 0.0652 | 0.2907 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2877C>A | H959Q 2D AIThe SynGAP1 missense variant H959Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely benign. Only ESM1b predicts a pathogenic outcome, representing the sole discordant signal. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H959Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | -8.657 | Likely Pathogenic | 0.126 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -0.77 | Neutral | 0.255 | Benign | 0.105 | Benign | 4.15 | Benign | 0.10 | Tolerated | 0.2264 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2877C>G | H959Q 2D AIThe SynGAP1 missense variant H959Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, while the single pathogenic prediction comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H959Q is most likely benign, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | -8.657 | Likely Pathogenic | 0.126 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -0.77 | Neutral | 0.255 | Benign | 0.105 | Benign | 4.15 | Benign | 0.10 | Tolerated | 0.2264 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2896C>T | H966Y 2D AIThe SynGAP1 missense variant H966Y is catalogued in gnomAD (6‑33443448‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all classify the change as benign or likely benign. Only polyPhen‑2 HumDiv reports a pathogenic prediction, representing the sole discordant signal. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. No Foldetta stability data are available, so folding‑stability evidence is inconclusive. Overall, the preponderance of predictions supports a benign classification for H966Y, and this conclusion is not contradicted by any ClinVar status (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | 6-33443448-C-T | -6.847 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -1.27 | Neutral | 0.878 | Possibly Damaging | 0.232 | Benign | 4.01 | Benign | 0.23 | Tolerated | 4.32 | 2 | 0.1465 | 0.4506 | 2 | 0 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||||||||
| c.3322A>G | S1108G 2D AIThe SynGAP1 missense variant S1108G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs three pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -6.496 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -2.59 | Deleterious | 0.568 | Possibly Damaging | 0.239 | Benign | 2.46 | Pathogenic | 0.16 | Tolerated | 0.2268 | 0.3975 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3418A>C | T1140P 2D AIThe SynGAP1 missense variant T1140P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for T1140P, and this conclusion is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.708094 | Binding | 0.293 | 0.854 | 1.000 | -1.921 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -1.49 | Neutral | 0.761 | Possibly Damaging | 0.478 | Possibly Damaging | 2.61 | Benign | 0.32 | Tolerated | 0.2044 | 0.3837 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3434A>G | N1145S 2D AIThe SynGAP1 missense variant N1145S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33444469‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, PolyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.722723 | Binding | 0.284 | 0.850 | 1.000 | Uncertain | 1 | 6-33444469-A-G | 2 | 1.24e-6 | -0.989 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.308 | Likely Benign | -1.15 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 5.55 | Benign | 0.89 | Tolerated | 4.32 | 4 | 0.4078 | 0.6708 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||||||||
| c.3448G>T | A1150S 2D AIThe SynGAP1 missense variant A1150S is reported in gnomAD (ID 6‑33444483‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.795712 | Binding | 0.371 | 0.831 | 0.625 | 6-33444483-G-T | 7 | 4.34e-6 | -2.656 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.49 | Neutral | 0.951 | Possibly Damaging | 0.752 | Possibly Damaging | 2.37 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.2656 | 0.5694 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||||
| c.3749A>T | Q1250L 2D AIThe SynGAP1 missense variant Q1250L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) and pathogenic predictions (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates “Likely Benign.” No Foldetta stability analysis is available for this residue. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar, which contains no pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.759478 | Disordered | 0.360484 | Uncertain | 0.881 | 0.554 | 0.750 | -4.409 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -3.49 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.65 | Benign | 0.02 | Affected | 0.0613 | 0.3946 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.37A>T | I13F 2D AIThe SynGAP1 missense variant I13F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.482657 | Uncertain | 0.318 | 0.916 | 0.375 | -3.359 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -0.09 | Neutral | 0.107 | Benign | 0.010 | Benign | 4.04 | Benign | 0.00 | Affected | 0.0654 | 0.3972 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.101A>T | Y34F 2D AIThe SynGAP1 missense variant Y34F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -3.275 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.50 | Neutral | 0.458 | Possibly Damaging | 0.481 | Possibly Damaging | 4.20 | Benign | 0.00 | Affected | 0.2604 | 0.3591 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.1429A>G | M477V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M477V is listed in ClinVar with no submitted interpretation and is present in the gnomAD database (variant ID 6‑33438461‑A‑G). Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen2_HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all predict benign or likely benign. Only two tools predict a pathogenic outcome: polyPhen2_HumDiv and FATHMM. Predictions from FoldX and Foldetta are uncertain. High‑accuracy methods reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, while Foldetta remains inconclusive. Taken together, the majority of evidence supports a benign classification for M477V, and this assessment does not contradict the ClinVar status, which currently has no pathogenic claim. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.268042 | Structured | 0.408680 | Uncertain | 0.761 | 0.250 | 0.000 | 6-33438461-A-G | 1 | 6.20e-7 | -3.995 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 1.64 | Ambiguous | 0.3 | 0.42 | Likely Benign | 1.03 | Ambiguous | 0.24 | Likely Benign | 0.209 | Likely Benign | -1.04 | Neutral | 0.716 | Possibly Damaging | 0.204 | Benign | -1.19 | Pathogenic | 0.22 | Tolerated | 3.37 | 34 | 0.3093 | 0.3445 | 1 | 2 | 2.3 | -32.06 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||
| c.1963C>G | L655V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L655V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. No tools predict pathogenicity, and the uncertain stability assessment does not alter the overall benign inference. Therefore, based on the available predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.015344 | Structured | 0.268808 | Uncertain | 0.961 | 0.274 | 0.000 | -6.743 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.78 | Ambiguous | 0.0 | 0.58 | Ambiguous | 0.68 | Ambiguous | 0.38 | Likely Benign | 0.058 | Likely Benign | -0.62 | Neutral | 0.008 | Benign | 0.003 | Benign | 3.45 | Benign | 0.44 | Tolerated | 0.1551 | 0.3190 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2404G>T | G802C 2D AIThe SynGAP1 missense variant G802C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence—including the consensus of multiple independent predictors and the high‑accuracy tools—supports a benign classification for G802C. This conclusion is consistent with the lack of any ClinVar pathogenic annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.894241 | Disordered | 0.681966 | Binding | 0.294 | 0.898 | 0.625 | -6.332 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.127 | Likely Benign | -1.60 | Neutral | 0.983 | Probably Damaging | 0.819 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 0.1426 | 0.4126 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||
| c.2702C>G | A901G 2D AIThe SynGAP1 missense variant A901G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of available predictions points to a benign impact, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.489838 | Uncertain | 0.306 | 0.917 | 0.375 | -3.928 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -1.52 | Neutral | 0.622 | Possibly Damaging | 0.165 | Benign | 2.61 | Benign | 0.37 | Tolerated | 0.2030 | 0.5188 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2734A>T | T912S 2D AIThe SynGAP1 missense variant T912S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for T912S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.740671 | Binding | 0.285 | 0.909 | 0.250 | -2.647 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -1.05 | Neutral | 0.994 | Probably Damaging | 0.856 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.3078 | 0.3893 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2735C>G | T912S 2D AIThe SynGAP1 missense variant T912S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for T912S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.740671 | Binding | 0.285 | 0.909 | 0.250 | -2.647 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.05 | Neutral | 0.994 | Probably Damaging | 0.856 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.3078 | 0.3893 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.611C>G | S204C 2D 3DClick to see structure in 3D Viewer AISynGAP1 S204C is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, Foldetta, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. FoldX and Rosetta individually report uncertain stability changes. Overall, the majority of computational evidence supports a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.268042 | Structured | 0.420667 | Uncertain | 0.816 | 0.405 | 0.125 | Uncertain | 1 | -6.613 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.4 | -1.13 | Ambiguous | -0.24 | Likely Benign | 0.10 | Likely Benign | 0.148 | Likely Benign | -0.64 | Neutral | 0.978 | Probably Damaging | 0.753 | Possibly Damaging | 4.13 | Benign | 0.05 | Affected | 3.44 | 10 | 0.0665 | 0.5237 | 0 | -1 | 3.3 | 16.06 | 223.6 | -13.8 | 0.6 | 0.3 | 0.0 | 0.2 | X | Uncertain | The hydroxyl-containing Ser204, located in the N-terminal loop before the first anti-parallel β sheet strand (res. Ile205-Pro208), is replaced by the thiol-containing cysteine. In the WT simulations, Ser204 simultaneously forms hydrogen bonds with the backbone carbonyl of Asp201 and the hydroxyl group of Thr224, helping to stabilize the two anti-parallel β strands (res. Ile205-Lys207 and Cys219-Thr223) at the end of the β sheet. Since the thiol group of cysteine forms weaker hydrogen bonds than the hydroxyl group of serine, Cys204 does not maintain the hydrogen bond network as stably as Ser204 in the variant simulations. However, because the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | ||||||||||||||||
| c.2329C>A | L777I 2D AIThe SynGAP1 missense variant L777I is listed in gnomAD (ID 6‑33442487‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which reports “Likely Benign.” Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | 6-33442487-C-A | -5.346 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.096 | Likely Benign | -0.85 | Neutral | 0.843 | Possibly Damaging | 0.920 | Probably Damaging | 4.05 | Benign | 0.02 | Affected | 3.64 | 6 | 0.1041 | 0.4327 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||
| c.2651G>A | R884Q 2D AIThe SynGAP1 missense variant R884Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443203‑G‑A). All available in silico predictors agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not reported, so they are unavailable. Overall, the computational evidence strongly supports a benign classification, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.641526 | Binding | 0.305 | 0.898 | 0.250 | Uncertain | 2 | 6-33443203-G-A | 5 | 3.10e-6 | -3.785 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.42 | Neutral | 0.012 | Benign | 0.004 | Benign | 2.62 | Benign | 0.36 | Tolerated | 4.32 | 4 | 0.2874 | 0.2332 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.3259T>A | S1087T 2D AIThe SynGAP1 missense variant S1087T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | -4.455 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -1.01 | Neutral | 0.790 | Possibly Damaging | 0.266 | Benign | 2.66 | Benign | 0.25 | Tolerated | 0.1333 | 0.6503 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3337G>T | G1113C 2D AIThe SynGAP1 missense variant G1113C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -7.917 | In-Between | 0.128 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.64 | Neutral | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 2.50 | Benign | 0.06 | Tolerated | 0.1325 | 0.4956 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3977C>A | P1326Q 2D AIThe SynGAP1 missense variant P1326Q is listed in ClinVar (ID 2806103.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33451851‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar “Uncertain” classification, which remains inconclusive. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.887377 | Binding | 0.393 | 0.782 | 0.875 | Uncertain | 1 | 6-33451851-C-A | 1 | 6.40e-7 | -5.422 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.138 | Likely Benign | -0.86 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 3.62 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1769 | 0.4457 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||
| c.46A>G | M16V 2D AIThe SynGAP1 missense variant M16V is reported in gnomAD (ID 6‑33420310‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for M16V, and this conclusion does not contradict any ClinVar classification, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.440853 | Structured | 0.459925 | Uncertain | 0.346 | 0.908 | 0.375 | 6-33420310-A-G | 16 | 1.05e-5 | -1.595 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -0.07 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.30 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3561 | 0.3937 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||||||
| c.58C>G | P20A 2D AIThe SynGAP1 missense variant P20A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect for P20A, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.442804 | Uncertain | 0.448 | 0.899 | 0.500 | -3.120 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.31 | Neutral | 0.805 | Possibly Damaging | 0.539 | Possibly Damaging | 4.31 | Benign | 0.00 | Affected | 0.3753 | 0.5520 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.107A>T | H36L 2D AIThe SynGAP1 missense variant H36L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign classification. AlphaMissense‑Optimized also reports benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of evidence points to a benign effect. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -2.403 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -1.73 | Neutral | 0.010 | Benign | 0.011 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1310 | 0.6111 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.1178G>C | G393A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G393A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictions indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -4.507 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 1.93 | Ambiguous | 0.5 | -0.68 | Ambiguous | 0.63 | Ambiguous | 0.22 | Likely Benign | 0.381 | Likely Benign | -1.89 | Neutral | 0.176 | Benign | 0.039 | Benign | 1.33 | Pathogenic | 0.08 | Tolerated | 0.4143 | 0.5193 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1298C>T | A433V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A433V missense variant is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438203‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and Foldetta as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and therefore unavailable as evidence. Overall, the majority of predictions support a benign impact, and this is consistent with the lack of ClinVar pathogenic classification. Thus, the variant is most likely benign based on current computational evidence, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | 6-33438203-C-T | 240 | 1.49e-4 | -8.200 | Likely Pathogenic | 0.129 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.1 | -0.07 | Likely Benign | 0.21 | Likely Benign | 0.48 | Likely Benign | 0.096 | Likely Benign | -2.91 | Deleterious | 0.994 | Probably Damaging | 0.527 | Possibly Damaging | 3.43 | Benign | 0.04 | Affected | 3.37 | 29 | 0.0753 | 0.5104 | 0 | 0 | 2.4 | 28.05 | 214.5 | -45.8 | 0.0 | 0.0 | 0.0 | 0.2 | X | Potentially Benign | The methyl group of Ala433, located on the outer surface of an α helix (res. Met414-Glu436), does not interact with any nearby residues in the WT simulations. In the variant simulations, the iso-propyl side chain of Val433, which has a similarly hydrophobic profile as alanine, also does not form any lasting interactions at the helix surface. Accordingly, the residue swap does not negatively affect the protein structure based on the simulations. | ||||||||||||||||
| c.1300G>A | V434I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434I (ClinVar ID 212346.0, status Uncertain) is present in gnomAD (ID 6‑33438205‑G‑A). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No predictions or stability results are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, which does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | Uncertain | 1 | 6-33438205-G-A | 1 | 6.19e-7 | -6.999 | Likely Benign | 0.129 | Likely Benign | Likely Benign | -0.04 | Likely Benign | 0.0 | 0.22 | Likely Benign | 0.09 | Likely Benign | 0.31 | Likely Benign | 0.192 | Likely Benign | -0.82 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.53 | Benign | 0.18 | Tolerated | 3.37 | 29 | 0.0675 | 0.3415 | 4 | 3 | 0.3 | 14.03 | 246.7 | -27.7 | 0.0 | 0.0 | 0.1 | 0.0 | X | Potentially Benign | The iso-propyl side chain of Val434, located at the end of an α helix (res. Met414-Glu436), packs against hydrophobic residues in an interhelix space (e.g., Met430, Ala707, Leu711). In the variant simulations, the sec-butyl group of Ile434 is able to form the same hydrophobic interactions. Accordingly, the residue swap does not negatively affect the protein structure based on the simulations. | |||||||||||||
| c.2243T>C | L748P 2D AIThe SynGAP1 missense variant L748P is reported in gnomAD (ID 6‑33441708‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | 6-33441708-T-C | 1 | 6.20e-7 | -2.732 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.69 | Neutral | 0.912 | Possibly Damaging | 0.611 | Possibly Damaging | 2.69 | Benign | 0.02 | Affected | 4.32 | 2 | 0.3503 | 0.1399 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||
| c.2398G>T | G800C 2D AIThe SynGAP1 missense variant G800C is not reported in ClinVar and has no allele in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a benign outcome; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of a ClinVar pathogenic annotation. Based on the aggregate predictions, the variant is most likely benign, and this is consistent with the lack of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.852992 | Disordered | 0.588350 | Binding | 0.303 | 0.884 | 0.625 | -7.074 | In-Between | 0.129 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -1.53 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 2.70 | Benign | 0.15 | Tolerated | 0.1102 | 0.4189 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||
| c.2869C>T | H957Y 2D AIThe SynGAP1 missense variant H957Y is listed in gnomAD (ID 6‑33443421‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which reports “Likely Benign.” Pathogenic predictions come from polyPhen‑2 HumDiv and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.968874 | Binding | 0.362 | 0.915 | 0.750 | 6-33443421-C-T | 1 | 6.20e-7 | -6.631 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -1.00 | Neutral | 0.510 | Possibly Damaging | 0.147 | Benign | 2.42 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.1560 | 0.4906 | 2 | 0 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||||||
| c.299A>C | Y100S 2D  AIThe SynGAP1 missense variant Y100S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -1.217 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -0.14 | Neutral | 0.675 | Possibly Damaging | 0.175 | Benign | 4.31 | Benign | 0.00 | Affected | 0.5066 | 0.1600 | Weaken | -3 | -2 | 0.5 | -76.10 | ||||||||||||||||||||||||||||||||||||||
| c.3032G>T | G1011V 2D AIThe SynGAP1 missense variant G1011V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | -4.883 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -1.21 | Neutral | 0.473 | Possibly Damaging | 0.192 | Benign | 2.71 | Benign | 0.01 | Affected | 0.1352 | 0.3434 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3241G>A | A1081T 2D AIThe SynGAP1 missense variant A1081T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -3.887 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.07 | Neutral | 0.440 | Benign | 0.184 | Benign | 4.01 | Benign | 0.15 | Tolerated | 0.1567 | 0.6112 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3283C>G | P1095A 2D AIThe SynGAP1 missense variant P1095A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | -4.555 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.80 | Neutral | 0.580 | Possibly Damaging | 0.242 | Benign | 2.79 | Benign | 0.18 | Tolerated | 0.3122 | 0.5703 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.4025A>G | D1342G 2D  AIThe SynGAP1 missense variant D1342G is reported in gnomAD (variant ID 6‑33451899‑A‑G) but has no ClinVar entry. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.921076 | Disordered | 0.981682 | Binding | 0.316 | 0.678 | 0.875 | 6-33451899-A-G | 1 | 7.29e-7 | -3.227 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.021 | Likely Benign | -0.89 | Neutral | 0.225 | Benign | 0.045 | Benign | 4.05 | Benign | 0.09 | Tolerated | 4.32 | 4 | 0.3633 | 0.5601 | -1 | 1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||
| c.10T>A | S4T 2D AIThe SynGAP1 missense variant S4T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, all of which are benign, and therefore SGM‑Consensus also predicts benign. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.598 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.01 | Neutral | 0.140 | Benign | 0.153 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1383 | 0.6572 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1165T>C | S389P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S389P is listed in gnomAD (variant ID 6‑33438070‑T‑C) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.703578 | Disordered | 0.417444 | Uncertain | 0.306 | 0.803 | 0.875 | 6-33438070-T-C | -5.286 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 1.85 | Ambiguous | 2.0 | -0.99 | Ambiguous | 0.43 | Likely Benign | 0.16 | Likely Benign | 0.460 | Likely Benign | -0.44 | Neutral | 0.676 | Possibly Damaging | 0.693 | Possibly Damaging | 5.05 | Benign | 0.01 | Affected | 4.32 | 8 | 0.3098 | 0.7002 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||
| c.16G>A | A6T 2D AIThe SynGAP1 missense variant A6T is reported in gnomAD (variant ID 6-33420280‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus also reports likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.566480 | Disordered | 0.549054 | Binding | 0.377 | 0.920 | 0.875 | 6-33420280-G-A | -3.711 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.13 | Neutral | 0.027 | Benign | 0.004 | Benign | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1521 | 0.6836 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||||
| c.2155A>C | N719H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only two tools—polyPhen‑2 HumDiv and polyPhen‑2 HumVar—predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports Benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates Benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -6.310 | Likely Benign | 0.130 | Likely Benign | Likely Benign | -0.04 | Likely Benign | 0.0 | -0.36 | Likely Benign | -0.20 | Likely Benign | 0.12 | Likely Benign | 0.095 | Likely Benign | -2.22 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.71 | Benign | 0.19 | Tolerated | 0.0782 | 0.4209 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.2871T>A | H957Q 2D AIThe SynGAP1 missense variant H957Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.968874 | Binding | 0.362 | 0.915 | 0.750 | -6.304 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.87 | Neutral | 0.255 | Benign | 0.105 | Benign | 2.54 | Benign | 0.56 | Tolerated | 0.2227 | 0.4114 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2871T>G | H957Q 2D AIThe SynGAP1 missense variant H957Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.968874 | Binding | 0.362 | 0.915 | 0.750 | -6.304 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.87 | Neutral | 0.255 | Benign | 0.105 | Benign | 2.54 | Benign | 0.56 | Tolerated | 0.2227 | 0.4114 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2886C>A | H962Q 2D AIThe SynGAP1 missense variant H962Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.984483 | Binding | 0.369 | 0.886 | 0.750 | -8.161 | Likely Pathogenic | 0.130 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -1.04 | Neutral | 0.325 | Benign | 0.045 | Benign | 4.19 | Benign | 0.06 | Tolerated | 0.1943 | 0.3844 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2886C>G | H962Q 2D AIThe SynGAP1 missense variant H962Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. There is no ClinVar entry to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.984483 | Binding | 0.369 | 0.886 | 0.750 | -8.161 | Likely Pathogenic | 0.130 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -1.04 | Neutral | 0.325 | Benign | 0.045 | Benign | 4.19 | Benign | 0.06 | Tolerated | 0.1943 | 0.3844 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3848C>G | P1283R 2D AIThe SynGAP1 missense variant P1283R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.819686 | Binding | 0.484 | 0.732 | 0.875 | -3.643 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.40 | Neutral | 0.911 | Possibly Damaging | 0.567 | Possibly Damaging | 2.72 | Benign | 0.05 | Affected | 0.1377 | 0.2211 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.591G>C | E197D 2D AIThe SynGAP1 missense variant E197D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions strongly supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | 0.784 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.066 | Likely Benign | 1.81 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.66 | Benign | 1.00 | Tolerated | 0.1579 | 0.3345 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.591G>T | E197D 2D AIThe SynGAP1 missense variant E197D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions strongly supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | 0.784 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.066 | Likely Benign | 1.81 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.66 | Benign | 1.00 | Tolerated | 0.1579 | 0.3345 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1033C>G | L345V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L345V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, Rosetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans toward benign. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -6.594 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 1.31 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.75 | Ambiguous | 0.92 | Ambiguous | 0.126 | Likely Benign | -1.96 | Neutral | 0.802 | Possibly Damaging | 0.277 | Benign | 1.77 | Pathogenic | 0.06 | Tolerated | 0.1411 | 0.3455 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1151G>T | G384V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G384V is catalogued in gnomAD (ID 6‑33438056‑G‑T) but has no entry in ClinVar. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a pathogenic effect on protein stability. Overall, the majority of evidence points to a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | 6-33438056-G-T | -6.968 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 3.69 | Destabilizing | 0.4 | 6.77 | Destabilizing | 5.23 | Destabilizing | -0.11 | Likely Benign | 0.460 | Likely Benign | -1.01 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 4.32 | 2 | 0.1599 | 0.3708 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||
| c.1934T>A | F645Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F645Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools with inconclusive results are FoldX (Uncertain) and premPS (Uncertain). High‑accuracy assessments show AlphaMissense‑Optimized predicting Benign, the SGM‑Consensus indicating Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicting Benign. No tool predicts pathogenicity. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as the variant is not currently classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -3.544 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.2 | 0.21 | Likely Benign | 0.37 | Likely Benign | -0.61 | Ambiguous | 0.141 | Likely Benign | 2.40 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.61 | Benign | 1.00 | Tolerated | 0.1506 | 0.2189 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.2222C>G | P741R 2D AIThe SynGAP1 missense variant P741R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -4.434 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -1.19 | Neutral | 0.642 | Possibly Damaging | 0.393 | Benign | 2.85 | Benign | 0.02 | Affected | 0.1303 | 0.2712 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2645G>C | G882A 2D AIThe SynGAP1 missense variant G882A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, whereas only FATHMM predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence indicates the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -3.876 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.05 | Neutral | 0.004 | Benign | 0.002 | Benign | 2.14 | Pathogenic | 0.79 | Tolerated | 0.3345 | 0.5268 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2816C>A | P939Q 2D AIThe SynGAP1 missense variant P939Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus remains inconclusive and Foldetta is unavailable. Overall, five of the nine evaluated tools predict pathogenicity versus four predicting benignity, giving a slight tilt toward a pathogenic interpretation. This prediction does not contradict ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.950 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -3.45 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.1551 | 0.4342 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3533A>T | Y1178F 2D AIThe SynGAP1 missense variant Y1178F is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta results are unavailable. Based on the consensus of all available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.568433 | Binding | 0.554 | 0.688 | 0.250 | -3.081 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.162 | Likely Benign | -0.64 | Neutral | 0.012 | Benign | 0.017 | Benign | 5.44 | Benign | 0.24 | Tolerated | 0.1893 | 0.2758 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.3904T>A | L1302M 2D AIThe SynGAP1 missense variant L1302M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.889642 | Binding | 0.429 | 0.842 | 0.875 | -5.246 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -1.34 | Neutral | 0.960 | Probably Damaging | 0.799 | Possibly Damaging | 1.54 | Pathogenic | 0.00 | Affected | 0.0929 | 0.3439 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3979C>T | P1327S 2D AIThe SynGAP1 missense variant P1327S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33451853‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.900145 | Binding | 0.369 | 0.777 | 0.875 | Uncertain | 1 | 6-33451853-C-T | -4.744 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.092 | Likely Benign | 0.28 | Neutral | 0.980 | Probably Damaging | 0.857 | Possibly Damaging | 4.25 | Benign | 0.71 | Tolerated | 3.77 | 5 | 0.3098 | 0.4025 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.4022C>T | A1341V 2D AIThe SynGAP1 missense variant A1341V is catalogued in gnomAD (ID 6‑33451896‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes, while the single pathogenic signal comes from SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign likelihood. Foldetta results are unavailable, so they do not influence the assessment. Overall, the preponderance of evidence points to a benign impact for A1341V, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.980111 | Binding | 0.383 | 0.696 | 1.000 | 6-33451896-C-T | 1 | 6.29e-7 | -3.687 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -1.90 | Neutral | 0.006 | Benign | 0.011 | Benign | 4.04 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1398 | 0.6557 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.55G>A | A19T 2D AIThe SynGAP1 missense variant A19T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | -3.422 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -0.02 | Neutral | 0.371 | Benign | 0.036 | Benign | 4.14 | Benign | 0.00 | Affected | 0.2357 | 0.7248 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2026A>T | S676C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen2_HumVar, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen2_HumDiv, SIFT, and ESM1b. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Based on the overall consensus of the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -9.230 | Likely Pathogenic | 0.132 | Likely Benign | Likely Benign | 0.47 | Likely Benign | 0.1 | 0.77 | Ambiguous | 0.62 | Ambiguous | 0.15 | Likely Benign | 0.164 | Likely Benign | -2.45 | Neutral | 0.959 | Probably Damaging | 0.431 | Benign | 3.35 | Benign | 0.01 | Affected | 0.1113 | 0.6352 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.2872C>T | H958Y 2D AIThe SynGAP1 missense variant H958Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and ESM1b—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.976011 | Binding | 0.371 | 0.913 | 0.750 | -8.393 | Likely Pathogenic | 0.132 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.03 | Neutral | 0.836 | Possibly Damaging | 0.232 | Benign | 4.14 | Benign | 0.06 | Tolerated | 0.1792 | 0.4706 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2900G>C | R967P 2D AIThe SynGAP1 missense variant R967P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for R967P, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | -2.506 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -0.76 | Neutral | 0.996 | Probably Damaging | 0.828 | Possibly Damaging | 4.14 | Benign | 0.17 | Tolerated | 0.2145 | 0.5533 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.313T>A | S105T 2D AIThe SynGAP1 missense variant S105T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a benign effect. AlphaMissense‑Optimized independently scores the variant as benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | -4.166 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.54 | Neutral | 0.012 | Benign | 0.007 | Benign | 4.06 | Benign | 0.00 | Affected | 0.1583 | 0.5212 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3140C>T | S1047L 2D AIThe SynGAP1 missense variant S1047L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443692‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta stability analysis is not available for this variant. Overall, the preponderance of computational evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | Uncertain | 1 | 6-33443692-C-T | 1 | 6.20e-7 | -4.062 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -0.63 | Neutral | 0.144 | Benign | 0.058 | Benign | 2.60 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1575 | 0.5555 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.3172G>T | G1058C 2D AIThe SynGAP1 missense variant G1058C is reported in gnomAD (ID 6‑33443724‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and ESM1b. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | 6-33443724-G-T | 1 | 6.21e-7 | -9.384 | Likely Pathogenic | 0.132 | Likely Benign | Likely Benign | 0.264 | Likely Benign | -0.19 | Neutral | 0.600 | Possibly Damaging | 0.433 | Benign | 5.19 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1456 | 0.4627 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.3248A>G | K1083R 2D AIThe SynGAP1 missense variant K1083R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -2.212 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -0.53 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 4.06 | Benign | 0.84 | Tolerated | 0.5053 | 0.1903 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.3881C>G | A1294G 2D AIThe SynGAP1 missense variant A1294G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy predictors (AlphaMissense‑Optimized) indicate a benign outcome, while the consensus of other tools is split. Thus, the variant is most likely benign based on current predictions, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | -3.242 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.201 | Likely Benign | -3.06 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.1980 | 0.2695 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.4028A>T | H1343L 2D AIThe SynGAP1 missense variant H1343L is reported in gnomAD (ID 6‑33451902‑A‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.983646 | Binding | 0.350 | 0.677 | 0.875 | 6-33451902-A-T | -1.552 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -1.28 | Neutral | 0.053 | Benign | 0.012 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1222 | 0.5869 | -3 | -2 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||
| c.1172G>T | G391V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391V is listed in ClinVar as Benign (ClinVar ID 1014488.0) and is present in gnomAD (variant ID 6‑33438077‑G‑T). Prediction tools that classify the variant as benign include premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. Tools that predict pathogenicity are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall prediction leans toward a benign effect. This conclusion aligns with the ClinVar benign classification, so there is no contradiction with the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | Likely Benign | 1 | 6-33438077-G-T | 3 | 1.86e-6 | -6.642 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 4.23 | Destabilizing | 1.3 | 4.81 | Destabilizing | 4.52 | Destabilizing | -0.11 | Likely Benign | 0.595 | Likely Pathogenic | -0.98 | Neutral | 0.994 | Probably Damaging | 0.887 | Possibly Damaging | 1.32 | Pathogenic | 0.10 | Tolerated | 3.69 | 8 | 0.1621 | 0.3821 | -1 | -3 | 4.6 | 42.08 | 228.6 | -69.0 | 0.0 | 0.8 | -0.5 | 0.3 | Uncertain | Gly387 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364 and res. Ala399-Ile411). The Ω loop is assumed to directly interact with the membrane, and it is observed to move arbitrarily throughout the WT solvent simulations. This loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play significant roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are visualized in the variant’s simulations, Val391 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. Since the effects on the Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.2207G>T | R736L 2D AIThe SynGAP1 missense variant R736L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | -4.173 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -1.27 | Neutral | 0.653 | Possibly Damaging | 0.361 | Benign | 2.60 | Benign | 0.00 | Affected | 0.1856 | 0.3180 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2224C>T | R742W 2D  AIThe SynGAP1 missense variant R742W is listed in ClinVar (ID 2581888.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441689‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | Uncertain | 1 | 6-33441689-C-T | 6 | 3.72e-6 | -7.725 | In-Between | 0.133 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -1.71 | Neutral | 0.992 | Probably Damaging | 0.684 | Possibly Damaging | 2.66 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1638 | 0.2839 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||
| c.2413C>A | L805M 2D AIThe SynGAP1 missense variant L805M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | -4.229 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -0.67 | Neutral | 0.029 | Benign | 0.033 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 0.0911 | 0.3867 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||
| c.2893C>T | H965Y 2D AIThe SynGAP1 missense variant H965Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. The consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No tool predicts pathogenicity, and the single uncertain result from ESM1b does not alter the overall benign consensus. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta data are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -7.121 | In-Between | 0.133 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -1.02 | Neutral | 0.327 | Benign | 0.147 | Benign | 4.03 | Benign | 0.25 | Tolerated | 0.1582 | 0.4906 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3238G>A | A1080T 2D AIThe SynGAP1 missense variant A1080T (ClinVar ID 1473274.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33443790‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly suggests the variant is most likely benign, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | Conflicting | 2 | 6-33443790-G-A | 17 | 1.06e-5 | -3.928 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.19 | Neutral | 0.253 | Benign | 0.042 | Benign | 4.10 | Benign | 0.60 | Tolerated | 3.77 | 5 | 0.1564 | 0.7103 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3259T>C | S1087P 2D AIThe SynGAP1 missense variant S1087P is reported in gnomAD (ID 6‑33443811‑T‑C) and has no ClinVar entry. All available in silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Based on the unanimous benign predictions and lack of ClinVar pathogenic annotation, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | 6-33443811-T-C | 1 | 6.34e-7 | -2.946 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.135 | Likely Benign | -1.92 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.56 | Benign | 0.12 | Tolerated | 3.77 | 5 | 0.1803 | 0.5700 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.823C>G | P275A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P275A is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33437728‑C‑G). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments are less decisive: AlphaMissense‑Optimized reports a benign outcome, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is uncertain. Consequently, the overall evidence leans toward a benign interpretation, with no ClinVar record to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.059222 | Structured | 0.353469 | Uncertain | 0.811 | 0.208 | 0.250 | 6-33437728-C-G | 1 | 6.20e-7 | -6.137 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 1.87 | Ambiguous | 0.2 | 1.11 | Ambiguous | 1.49 | Ambiguous | 0.50 | Likely Benign | 0.410 | Likely Benign | -4.95 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.32 | Tolerated | 3.38 | 19 | 0.3475 | 0.3243 | -1 | 1 | 3.4 | -26.04 | |||||||||||||||||||||||||
| c.99A>C | Q33H 2D AIThe SynGAP1 missense variant Q33H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.436712 | Uncertain | 0.342 | 0.860 | 0.375 | -0.450 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.95 | Neutral | 0.704 | Possibly Damaging | 0.198 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1856 | 0.4509 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.99A>T | Q33H 2D AIThe SynGAP1 missense variant Q33H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.436712 | Uncertain | 0.342 | 0.860 | 0.375 | -0.450 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.95 | Neutral | 0.704 | Possibly Damaging | 0.198 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1856 | 0.4509 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.109T>A | S37T 2D AIThe SynGAP1 missense variant S37T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -3.854 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.60 | Neutral | 0.140 | Benign | 0.481 | Possibly Damaging | 3.95 | Benign | 0.00 | Affected | 0.2093 | 0.5974 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.213C>A | D71E 2D AIThe SynGAP1 missense variant D71E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.276 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.28 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.39 | Benign | 0.00 | Affected | 0.1490 | 0.5922 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.213C>G | D71E 2D AIThe SynGAP1 missense variant D71E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence or population data. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.276 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.28 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.39 | Benign | 0.00 | Affected | 0.1490 | 0.5922 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2249G>T | G750V 2D AIThe SynGAP1 missense variant G750V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show AlphaMissense‑Optimized as benign, while the remaining high‑confidence tools (PROVEAN, polyPhen‑2, SIFT, FATHMM) all indicate pathogenicity. Overall, the majority of evidence (five pathogenic vs four benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -4.512 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.54 | Deleterious | 0.984 | Probably Damaging | 0.850 | Possibly Damaging | 2.45 | Pathogenic | 0.01 | Affected | 0.1270 | 0.4212 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2278A>C | M760L 2D AIThe SynGAP1 missense variant M760L is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts a pathogenic outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.260 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -0.68 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.69 | Benign | 0.15 | Tolerated | 0.1802 | 0.4805 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2278A>T | M760L 2D AIThe SynGAP1 missense variant M760L is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts a pathogenic outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.260 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -0.68 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.69 | Benign | 0.15 | Tolerated | 0.1802 | 0.4805 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2329C>G | L777V 2D AIThe SynGAP1 missense variant L777V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | -5.693 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -1.05 | Neutral | 0.843 | Possibly Damaging | 0.920 | Probably Damaging | 4.07 | Benign | 0.05 | Affected | 0.1651 | 0.3977 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2594C>T | A865V 2D AIThe SynGAP1 missense variant A865V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only polyPhen‑2 HumDiv predicts it as pathogenic. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the computational evidence overwhelmingly suggests that A865V is most likely benign, and this assessment is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.626222 | Binding | 0.271 | 0.788 | 0.250 | -4.639 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -1.42 | Neutral | 0.611 | Possibly Damaging | 0.419 | Benign | 2.70 | Benign | 0.37 | Tolerated | 0.1145 | 0.6031 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2629C>A | L877M 2D AIThe SynGAP1 missense variant L877M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the two polyPhen‑2 scores (HumDiv and HumVar) predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.653063 | Disordered | 0.634010 | Binding | 0.265 | 0.875 | 0.250 | -6.266 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.55 | Neutral | 0.922 | Possibly Damaging | 0.776 | Possibly Damaging | 2.58 | Benign | 0.15 | Tolerated | 0.0813 | 0.3532 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3758C>G | A1253G 2D AIThe SynGAP1 missense variant A1253G is predicted to be benign by all evaluated in silico tools. Consensus predictions from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized uniformly indicate a benign effect. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a likely benign outcome. No Foldetta stability analysis is available for this residue. The variant is not listed in ClinVar and has no entry in gnomAD, so there is no external evidence to contradict the computational predictions. Based on the collective predictions, the variant is most likely benign, and this assessment aligns with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.661982 | Disordered | 0.391377 | Uncertain | 0.881 | 0.550 | 0.750 | -4.772 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -1.23 | Neutral | 0.151 | Benign | 0.148 | Benign | 2.76 | Benign | 0.26 | Tolerated | 0.1688 | 0.3407 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.385T>G | S129A 2D AIThe SynGAP1 missense variant S129A is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors—including REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as benign. No tool predicts pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.713635 | Binding | 0.311 | 0.880 | 0.625 | -3.388 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.090 | Likely Benign | -0.08 | Neutral | 0.007 | Benign | 0.007 | Benign | 4.21 | Benign | 0.15 | Tolerated | 0.5031 | 0.4619 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.4026C>A | D1342E 2D AIThe SynGAP1 missense variant D1342E is catalogued in gnomAD (ID 6‑33451900‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes. Only SIFT predicts a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that D1342E is most likely benign, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.921076 | Disordered | 0.981682 | Binding | 0.316 | 0.678 | 0.875 | 6-33451900-C-A | -3.169 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.88 | Neutral | 0.225 | Benign | 0.084 | Benign | 4.16 | Benign | 0.04 | Affected | 4.32 | 4 | 0.2130 | 0.5720 | 2 | 3 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||
| c.4026C>G | D1342E 2D AIThe SynGAP1 missense variant D1342E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions points to a benign effect for D1342E, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.921076 | Disordered | 0.981682 | Binding | 0.316 | 0.678 | 0.875 | -3.169 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.88 | Neutral | 0.225 | Benign | 0.084 | Benign | 4.16 | Benign | 0.04 | Affected | 4.32 | 4 | 0.2130 | 0.5720 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.108T>A | H36Q 2D AIThe SynGAP1 missense variant H36Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -3.642 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.16 | Neutral | 0.182 | Benign | 0.046 | Benign | 4.34 | Benign | 0.00 | Affected | 0.2042 | 0.4572 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.108T>G | H36Q 2D AIThe SynGAP1 missense variant H36Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -3.642 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.16 | Neutral | 0.182 | Benign | 0.046 | Benign | 4.34 | Benign | 0.00 | Affected | 0.2042 | 0.4572 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.1090C>G | P364A 2D 3DClick to see structure in 3D Viewer AISynGAP1 P364A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Stability‑based methods (FoldX, Rosetta, premPS) give uncertain outcomes, and Foldetta is unavailable. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, does not provide a definitive result. Overall, the predictions are discordant; the majority of tools lean toward pathogenicity, but a substantial subset suggests benign. Based on the predictions, the variant is most likely pathogenic, and this does not contradict ClinVar status because there is no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.439474 | Uncertain | 0.942 | 0.590 | 0.250 | -8.077 | Likely Pathogenic | 0.135 | Likely Benign | Likely Benign | 0.79 | Ambiguous | 0.0 | 0.69 | Ambiguous | 0.74 | Ambiguous | 0.60 | Ambiguous | 0.320 | Likely Benign | -6.10 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.56 | Pathogenic | 0.06 | Tolerated | 0.3496 | 0.5186 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1150G>T | G384C 2D AIThe SynGAP1 missense variant G384C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic; and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split and is treated as unavailable. Overall, the majority of predictions (seven pathogenic vs. five benign) and the pathogenic Foldetta result indicate that the variant is most likely pathogenic, with no contradiction to ClinVar status because the variant is not yet classified there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | -8.363 | Likely Pathogenic | 0.135 | Likely Benign | Likely Benign | 2.41 | Destabilizing | 2.0 | 5.50 | Destabilizing | 3.96 | Destabilizing | 0.17 | Likely Benign | 0.456 | Likely Benign | -1.07 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1530 | 0.4240 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.2021C>A | T674N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome; ESM1b is uncertain and does not influence the consensus. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports Benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Benign. No contradictory evidence is present. **Based on the aggregate predictions, the variant is most likely benign, and this conclusion does not conflict with the ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -7.839 | In-Between | 0.135 | Likely Benign | Likely Benign | -0.21 | Likely Benign | 0.0 | 0.15 | Likely Benign | -0.03 | Likely Benign | 0.19 | Likely Benign | 0.119 | Likely Benign | -0.71 | Neutral | 0.958 | Probably Damaging | 0.671 | Possibly Damaging | 3.44 | Benign | 0.17 | Tolerated | 0.1263 | 0.4979 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.215G>A | R72Q 2D AIThe SynGAP1 missense variant R72Q is reported in gnomAD (variant ID 6-33425823‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R72Q, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.455349 | Uncertain | 0.355 | 0.819 | 0.375 | 6-33425823-G-A | -4.413 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -0.13 | Neutral | 0.829 | Possibly Damaging | 0.238 | Benign | 4.20 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3570 | 0.2380 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||||||
| c.2587C>A | L863M 2D AIThe SynGAP1 missense variant L863M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic effect. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta results are not available, so they do not influence the overall assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.594839 | Binding | 0.267 | 0.795 | 0.250 | -5.137 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -0.21 | Neutral | 0.999 | Probably Damaging | 0.990 | Probably Damaging | 4.04 | Benign | 0.28 | Tolerated | 0.0811 | 0.3922 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.26A>T | H9L 2D AIThe SynGAP1 missense variant H9L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | -2.603 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.151 | Likely Benign | 0.48 | Neutral | 0.024 | Benign | 0.002 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1255 | 0.6285 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2929G>C | A977P 2D AIThe SynGAP1 missense variant A977P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A977P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -2.665 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -1.12 | Neutral | 0.990 | Probably Damaging | 0.892 | Possibly Damaging | 3.94 | Benign | 0.02 | Affected | 0.2405 | 0.5212 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.292C>T | H98Y 2D AIThe SynGAP1 missense variant H98Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -4.050 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -0.86 | Neutral | 0.444 | Benign | 0.024 | Benign | 4.19 | Benign | 0.00 | Affected | 0.0950 | 0.5024 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3107A>C | Q1036P 2D AIThe SynGAP1 missense variant Q1036P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a damaging or pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple prediction algorithms and the consensus score indicates that the variant is most likely benign, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -2.491 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -2.00 | Neutral | 0.802 | Possibly Damaging | 0.238 | Benign | 2.57 | Benign | 0.01 | Affected | 0.2018 | 0.5564 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3779A>G | K1260R 2D AIThe SynGAP1 missense variant K1260R is catalogued in gnomAD (ID 6‑33446771‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; those that predict pathogenicity are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective predictions point to a benign impact, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.509769 | Disordered | 0.625808 | Binding | 0.890 | 0.575 | 0.250 | 6-33446771-A-G | 1 | 6.20e-7 | -6.033 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.254 | Likely Benign | -2.29 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3655 | 0.0878 | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||
| c.3860C>A | P1287H 2D AIThe SynGAP1 missense variant P1287H is recorded in gnomAD (6‑33447908‑C‑A) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is not available for this variant. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.813701 | Binding | 0.538 | 0.777 | 0.750 | 6-33447908-C-A | -3.606 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -1.36 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 2.80 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1306 | 0.3448 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.3874C>A | L1292I 2D AIThe SynGAP1 missense variant L1292I is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.882643 | Binding | 0.586 | 0.800 | 0.625 | -5.480 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.050 | Likely Benign | 0.71 | Neutral | 0.002 | Benign | 0.006 | Benign | 3.23 | Benign | 1.00 | Tolerated | 0.1101 | 0.3323 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.77G>C | G26A 2D AIThe SynGAP1 missense variant G26A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G26A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.438291 | Uncertain | 0.351 | 0.878 | 0.375 | -3.308 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.25 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 4.02 | Benign | 0.00 | Affected | 0.4093 | 0.5251 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.125C>T | P42L 2D AIThe SynGAP1 missense variant P42L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.781 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.87 | Neutral | 0.909 | Possibly Damaging | 0.927 | Probably Damaging | 4.19 | Benign | 0.00 | Affected | 0.2312 | 0.5560 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1528A>G | I510V 2D 3DClick to see structure in 3D Viewer AISynGAP1 I510V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Pathogenic predictions arise from polyPhen‑2 HumDiv, SIFT, FATHMM, and premPS. FoldX and Rosetta analyses are inconclusive, and Foldetta stability assessment is unavailable. High‑accuracy methods reinforce the benign trend: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, while Foldetta provides no definitive result. Overall, the majority of evidence points to a benign effect for I510V, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.025762 | Structured | 0.250630 | Uncertain | 0.945 | 0.273 | 0.000 | -6.072 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 1.45 | Ambiguous | 0.2 | 0.50 | Ambiguous | 0.98 | Ambiguous | 1.13 | Destabilizing | 0.461 | Likely Benign | -1.00 | Neutral | 0.792 | Possibly Damaging | 0.332 | Benign | -1.36 | Pathogenic | 0.02 | Affected | 0.0999 | 0.2291 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.1801G>T | A601S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A601S missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools—FoldX, Rosetta, Foldetta, and premPS—return uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie and thus inconclusive. Foldetta, which integrates FoldX‑MD and Rosetta outputs, also yields an uncertain result. Overall, more tools (six) predict pathogenicity than benign (three), and no ClinVar evidence contradicts this assessment. Therefore, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.008895 | Structured | 0.174517 | Uncertain | 0.955 | 0.156 | 0.000 | -11.248 | Likely Pathogenic | 0.136 | Likely Benign | Likely Benign | 0.79 | Ambiguous | 0.1 | 1.63 | Ambiguous | 1.21 | Ambiguous | 0.79 | Ambiguous | 0.541 | Likely Pathogenic | -2.99 | Deleterious | 0.983 | Probably Damaging | 0.993 | Probably Damaging | 2.56 | Benign | 0.01 | Affected | 0.2732 | 0.4730 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.2575A>T | S859C 2D AIThe SynGAP1 missense variant S859C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The high‑accuracy consensus (SGM‑Consensus) derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN reports the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.648219 | Disordered | 0.497075 | Uncertain | 0.288 | 0.819 | 0.375 | -8.268 | Likely Pathogenic | 0.136 | Likely Benign | Likely Benign | 0.195 | Likely Benign | -2.01 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.94 | Benign | 0.03 | Affected | 0.1046 | 0.6153 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2597T>G | V866G 2D AIThe SynGAP1 missense variant V866G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for V866G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.638070 | Binding | 0.266 | 0.788 | 0.250 | -1.519 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -2.30 | Neutral | 0.912 | Possibly Damaging | 0.993 | Probably Damaging | 2.69 | Benign | 0.07 | Tolerated | 0.2548 | 0.2157 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.25C>T | H9Y 2D AIThe SynGAP1 missense variant H9Y is reported in gnomAD (ID 6‑33420289‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus itself is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | 6-33420289-C-T | -3.833 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.14 | Neutral | 0.047 | Benign | 0.006 | Benign | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1227 | 0.5024 | 2 | 0 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||||||||
| c.2660C>A | P887H 2D AIThe SynGAP1 missense variant P887H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for P887H, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.602269 | Binding | 0.348 | 0.925 | 0.500 | -4.900 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -1.71 | Neutral | 0.977 | Probably Damaging | 0.777 | Possibly Damaging | 2.73 | Benign | 0.13 | Tolerated | 0.1236 | 0.3275 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.307G>T | G103C 2D AIThe SynGAP1 missense variant G103C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for G103C, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -5.681 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.12 | Neutral | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 0.1494 | 0.3767 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3879C>A | D1293E 2D AIThe SynGAP1 missense variant D1293E is reported in gnomAD (variant ID 6‑33447927‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, SIFT and FATHMM predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | 6-33447927-C-A | -2.627 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -1.86 | Neutral | 0.011 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1512 | 0.3425 | 2 | 3 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||
| c.3879C>G | D1293E 2D AIThe SynGAP1 missense variant D1293E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for D1293E, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -2.627 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.86 | Neutral | 0.011 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1512 | 0.3425 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.3959C>A | P1320H 2D AIThe SynGAP1 missense variant P1320H is listed in gnomAD (ID 6‑33451833‑C‑A) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.946297 | Binding | 0.510 | 0.833 | 0.750 | 6-33451833-C-A | 2 | 1.29e-6 | -6.296 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -1.05 | Neutral | 0.998 | Probably Damaging | 0.990 | Probably Damaging | 4.17 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1955 | 0.5461 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||
| c.3974C>A | P1325H 2D AIThe SynGAP1 missense variant P1325H is reported in gnomAD (variant ID 6‑33451848‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No result is available from the Foldetta stability analysis. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.893621 | Binding | 0.439 | 0.791 | 0.875 | 6-33451848-C-A | -6.970 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.056 | Likely Benign | 0.10 | Neutral | 0.704 | Possibly Damaging | 0.187 | Benign | 4.04 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2103 | 0.4364 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.511G>A | A171T 2D AIThe SynGAP1 missense variant A171T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT predicts pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign impact. This prediction does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.492272 | Uncertain | 0.358 | 0.652 | 0.375 | -2.388 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.019 | Likely Benign | -0.51 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.25 | Benign | 0.03 | Affected | 0.1075 | 0.5763 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.595A>C | N199H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No predictions are missing or inconclusive. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -6.387 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.1 | 0.15 | Likely Benign | 0.16 | Likely Benign | 0.20 | Likely Benign | 0.064 | Likely Benign | -1.99 | Neutral | 0.952 | Possibly Damaging | 0.496 | Possibly Damaging | 4.17 | Benign | 0.08 | Tolerated | 0.0879 | 0.5635 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.794A>G | K265R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K265R is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster into two groups: benign predictions include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. No prediction or stability result is missing or inconclusive. Based on the aggregate evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.209395 | Structured | 0.309758 | Uncertain | 0.936 | 0.275 | 0.000 | -3.366 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.1 | -0.44 | Likely Benign | -0.20 | Likely Benign | 0.30 | Likely Benign | 0.223 | Likely Benign | -1.08 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.90 | Pathogenic | 0.36 | Tolerated | 0.4713 | 0.0976 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.92G>A | R31Q 2D AIThe SynGAP1 missense variant R31Q is listed in ClinVar (ID 1977609.0) with an “Uncertain” status and is present in gnomAD (6‑33423501‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” classification and suggests the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.549308 | Disordered | 0.437905 | Uncertain | 0.324 | 0.878 | 0.250 | Uncertain | 1 | 6-33423501-G-A | 7 | 4.34e-6 | -4.434 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -0.92 | Neutral | 0.829 | Possibly Damaging | 0.614 | Possibly Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3605 | 0.3355 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.962G>A | R321H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R321H is listed in ClinVar as Benign (ClinVar ID 1940706) and is present in gnomAD (ID 6-33437867-G-A). Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools predicting a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM; premPS is reported as Uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, Foldetta as Benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence supports a benign effect, aligning with the ClinVar classification and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | Benign | 1 | 6-33437867-G-A | 8 | 4.96e-6 | -8.751 | Likely Pathogenic | 0.136 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.1 | -0.36 | Likely Benign | 0.06 | Likely Benign | 0.59 | Ambiguous | 0.323 | Likely Benign | -1.43 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.92 | Pathogenic | 0.25 | Tolerated | 3.38 | 23 | 0.2930 | 0.0936 | 2 | 0 | 1.3 | -19.05 | 218.5 | 86.9 | 1.1 | 0.0 | 0.3 | 0.0 | X | Potentially Benign | The guanidinium group of Arg321, located in a β hairpin loop linking two anti-parallel β sheet strands (res. Thr305-Asn315, res. Ala322-Asp330), faces outward without forming any stable interactions in the WT simulations. Similarly, in the variant simulations, the imidazole ring of His321 also points outward without making any stable intra-protein interactions. Thus, the residue swap does not seem to cause adverse effects on the protein structure based on the simulations. However, β hairpins are potential nucleation sites during the initial stages of protein folding, so even minor changes in them could be significant. | ||||||||||||||
| c.1198G>C | V400L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V400L is listed in ClinVar as Benign (ClinVar ID 1166313) and is present in gnomAD (6‑33438103‑G‑C). Prediction tools that agree on a benign effect include REVEL, FoldX‑MD (uncertain), Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.398279 | Structured | 0.415488 | Uncertain | 0.951 | 0.451 | 0.000 | Benign/Likely benign | 2 | 6-33438103-G-C | 22 | 1.36e-5 | -1.000 | Likely Benign | 0.137 | Likely Benign | Likely Benign | -0.71 | Ambiguous | 0.2 | 0.39 | Likely Benign | -0.16 | Likely Benign | -0.29 | Likely Benign | 0.325 | Likely Benign | -0.60 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.33 | Benign | 0.64 | Tolerated | 3.38 | 27 | 0.1006 | 0.5242 | 2 | 1 | -0.4 | 14.03 | 251.0 | -30.1 | 0.0 | 0.0 | 0.7 | 0.1 | X | Potentially Benign | The iso-propyl side chain of Val400, located in an anti-parallel β sheet strand (res. Ala399-Ile411), hydrophobically packs against hydrophobic residues within the anti-parallel β sheet of the C2 domain (e.g., Ile268, Ala404, Leu325, Leu402). Val400 is swapped for another hydrophobic residue, leucine, whose branched hydrocarbon side chain is of a comparable size and thus packs favorably within the C2 domain. In short, the residue swap has no apparent negative effect on the structure based on the variant simulations. | 10.1016/j.ajhg.2020.11.011 | ||||||||||||
| c.1198G>T | V400L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V400L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, premPS, AlphaMissense‑Default, AlphaMissense‑Optimized, and Rosetta. No tool predicts a pathogenic outcome; the only inconclusive result is from FoldX (Uncertain). High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Benign. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.398279 | Structured | 0.415488 | Uncertain | 0.951 | 0.451 | 0.000 | -1.000 | Likely Benign | 0.137 | Likely Benign | Likely Benign | -0.71 | Ambiguous | 0.2 | 0.39 | Likely Benign | -0.16 | Likely Benign | -0.29 | Likely Benign | 0.325 | Likely Benign | -0.60 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.33 | Benign | 0.64 | Tolerated | 3.38 | 27 | 0.1006 | 0.5242 | 2 | 1 | -0.4 | 14.03 | 251.0 | -30.1 | 0.0 | 0.0 | 0.7 | 0.1 | X | Potentially Benign | The iso-propyl side chain of Val400, located in an anti-parallel β sheet strand (res. Ala399-Ile411), hydrophobically packs against hydrophobic residues within the anti-parallel β sheet of the C2 domain (e.g., Ile268, Ala404, Leu325, Leu402). Val400 is swapped for another hydrophobic residue, leucine, whose branched hydrocarbon side chain is of a comparable size and thus packs favorably within the C2 domain. In short, the residue swap has no apparent negative effect on the structure based on the variant simulations. | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||
| c.1211C>G | A404G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A404G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS and SIFT. FoldX, Rosetta, and Foldetta give uncertain or inconclusive results and are treated as unavailable. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains uncertain. Overall, the majority of evidence points to a benign impact for A404G, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.232838 | Structured | 0.415505 | Uncertain | 0.965 | 0.355 | 0.000 | -5.277 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.92 | Ambiguous | 0.0 | 1.88 | Ambiguous | 1.40 | Ambiguous | 1.18 | Destabilizing | 0.099 | Likely Benign | -2.34 | Neutral | 0.045 | Benign | 0.013 | Benign | 4.03 | Benign | 0.00 | Affected | 0.2370 | 0.5054 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1228A>G | S410G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The remaining tools (FoldX, Rosetta, Foldetta, premPS, ESM1b) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -7.147 | In-Between | 0.137 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.1 | 1.33 | Ambiguous | 0.96 | Ambiguous | 0.85 | Ambiguous | 0.117 | Likely Benign | -2.54 | Deleterious | 0.952 | Possibly Damaging | 0.145 | Benign | 4.13 | Benign | 0.19 | Tolerated | 0.2651 | 0.5143 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||
| c.1909T>G | S637A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, SIFT, polyPhen‑2 (HumDiv and HumVar), REVEL, FoldX, and premPS. No tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is inconclusive (Uncertain). Taken together, the overwhelming majority of evidence indicates a benign effect. The variant’s status is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -4.186 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.1 | 0.99 | Ambiguous | 0.65 | Ambiguous | 0.22 | Likely Benign | 0.078 | Likely Benign | -0.64 | Neutral | 0.120 | Benign | 0.182 | Benign | 3.41 | Benign | 1.00 | Tolerated | 0.4853 | 0.2996 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.2570G>A | S857N 2D AIThe SynGAP1 missense variant S857N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.475747 | Uncertain | 0.288 | 0.826 | 0.375 | -5.065 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.128 | Likely Benign | 0.24 | Neutral | 0.991 | Probably Damaging | 0.982 | Probably Damaging | 4.09 | Benign | 1.00 | Tolerated | 0.1515 | 0.5034 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.70G>A | V24I 2D AIThe SynGAP1 missense variant V24I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6-33423479-G-A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of computational evidence supports a benign impact for V24I, and this conclusion does not contradict the ClinVar designation, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | Uncertain | 1 | 6-33423479-G-A | 9 | 5.58e-6 | -3.701 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -0.25 | Neutral | 0.043 | Benign | 0.031 | Benign | 3.96 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0844 | 0.4564 | 3 | 4 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||||
| c.1054A>C | T352P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T352P has no ClinVar entry and is not reported in gnomAD. Prediction tools that converge on a benign outcome include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, FATHMM, and Rosetta. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the balance of evidence favors a benign interpretation; this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | -3.562 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 1.04 | Ambiguous | 0.1 | 2.57 | Destabilizing | 1.81 | Ambiguous | 0.51 | Ambiguous | 0.159 | Likely Benign | -2.31 | Neutral | 0.627 | Possibly Damaging | 0.196 | Benign | 1.72 | Pathogenic | 0.20 | Tolerated | 0.2244 | 0.5968 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1910C>G | S637C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, FoldX, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; Foldetta predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence supports a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -10.040 | Likely Pathogenic | 0.138 | Likely Benign | Likely Benign | 0.25 | Likely Benign | 0.0 | 0.54 | Ambiguous | 0.40 | Likely Benign | 0.22 | Likely Benign | 0.169 | Likely Benign | -2.83 | Deleterious | 0.985 | Probably Damaging | 0.533 | Possibly Damaging | 3.34 | Benign | 0.01 | Affected | 0.1327 | 0.4439 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.2047A>G | I683V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683V is listed in ClinVar with an uncertain significance and is present in gnomAD (6‑33441306‑A‑G). Across a panel of in silico predictors, the majority indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (derived from a majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only polyPhen‑2 HumDiv classifies the change as pathogenic. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote) is benign, and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is inconclusive and therefore not considered evidence. No other tool provides a pathogenic signal. Consequently, the variant is most likely benign, and this assessment does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | Uncertain | 1 | 6-33441306-A-G | 2 | 1.24e-6 | -7.588 | In-Between | 0.138 | Likely Benign | Likely Benign | 0.90 | Ambiguous | 0.0 | 0.60 | Ambiguous | 0.75 | Ambiguous | 0.76 | Ambiguous | 0.112 | Likely Benign | -0.78 | Neutral | 0.538 | Possibly Damaging | 0.080 | Benign | 3.35 | Benign | 0.14 | Tolerated | 3.42 | 17 | 0.1021 | 0.2898 | 4 | 3 | -0.3 | -14.03 | 215.6 | 29.1 | 0.0 | 0.0 | -0.7 | 0.1 | X | Potentially Benign | The sec-butyl side chain of Ile683, located in an entangled α-α loop connecting the two α-helices (res. Ser641-Glu666 and res. Leu685-Val699), is sterically packed against His453 and Glu688. In the variant simulations, the iso-propyl side chain of Val683 has similar size and physicochemical properties as Ile630 in the WT, and thus, it is able to maintain similar interactions in the inter-helix space. Consequently, no negative structural effects are observed during the simulations due to the residue swap. | |||||||||||||
| c.2347A>C | M783L 2D AIThe SynGAP1 missense variant M783L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -1.915 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -0.48 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.99 | Benign | 1.00 | Tolerated | 0.1727 | 0.4461 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2347A>T | M783L 2D AIThe SynGAP1 missense variant M783L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify the change as benign. No tool in the dataset predicts pathogenicity. High‑accuracy consensus methods reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability approach, has no available result for this variant. Overall, the collective evidence strongly supports a benign interpretation, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -1.915 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.48 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.99 | Benign | 1.00 | Tolerated | 0.1727 | 0.4461 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2816C>T | P939L 2D AIThe SynGAP1 missense variant P939L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta are unavailable. Overall, five tools predict pathogenicity versus four predicting benign, so the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.191 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -3.69 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.2113 | 0.5142 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2930C>G | A977G 2D AIThe SynGAP1 missense variant A977G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A977G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -3.250 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.79 | Neutral | 0.965 | Probably Damaging | 0.702 | Possibly Damaging | 4.03 | Benign | 0.05 | Affected | 0.2180 | 0.4408 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3029T>A | F1010Y 2D AIThe SynGAP1 missense variant F1010Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | -3.297 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.96 | Neutral | 0.031 | Benign | 0.064 | Benign | 2.64 | Benign | 0.05 | Affected | 0.1651 | 0.2514 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3641G>A | R1214Q 2D AIThe SynGAP1 missense variant R1214Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33446633‑G‑A). Functional prediction tools cluster into two groups: the benign‑predicating set includes REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the pathogenic‑predicating set consists of polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Taken together, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, which is consistent with the ClinVar designation of uncertainty rather than a definitive pathogenic claim. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.497853 | Structured | 0.506868 | Binding | 0.903 | 0.566 | 0.375 | Uncertain | 1 | 6-33446633-G-A | 6 | 3.72e-6 | -6.059 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.69 | Neutral | 0.993 | Probably Damaging | 0.738 | Possibly Damaging | 2.65 | Benign | 0.02 | Affected | 3.77 | 5 | 0.2162 | 0.1530 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||||||
| c.371C>T | A124V 2D AIThe SynGAP1 A124V missense variant is listed in ClinVar (ID 1040523.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33432236‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of computational evidence indicates a benign effect, and this consensus does not contradict the ClinVar “Uncertain” designation. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.699139 | Binding | 0.340 | 0.883 | 0.750 | Conflicting | 2 | 6-33432236-C-T | 9 | 5.58e-6 | -4.259 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -1.52 | Neutral | 0.173 | Benign | 0.009 | Benign | 4.07 | Benign | 0.03 | Affected | 3.61 | 5 | 0.1415 | 0.7433 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||
| c.106C>T | H36Y 2D AIThe SynGAP1 missense variant H36Y is listed in ClinVar with an uncertain significance (ClinVar ID 2089635.0) and is present in the gnomAD database (gnomAD ID 6‑33423515‑C‑T). Functional prediction tools largely agree that the substitution is benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report a benign effect. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Benign. No Foldetta stability prediction is available. Overall, the computational evidence overwhelmingly supports a benign classification, which is consistent with the ClinVar designation of uncertain significance rather than a pathogenic claim. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | Uncertain | 1 | 6-33423515-C-T | 2 | 1.24e-6 | -3.461 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -1.03 | Neutral | 0.219 | Benign | 0.066 | Benign | 4.16 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1265 | 0.5024 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||||
| c.1429A>C | M477L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M477L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome; premPS is uncertain and therefore not counted as evidence. High‑accuracy assessments are consistent: AlphaMissense‑Optimized reports Benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Benign. Overall, the preponderance of evidence supports a benign classification for M477L, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.268042 | Structured | 0.408680 | Uncertain | 0.761 | 0.250 | 0.000 | -4.772 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.19 | Likely Benign | 0.15 | Likely Benign | 0.58 | Ambiguous | 0.236 | Likely Benign | -1.25 | Neutral | 0.000 | Benign | 0.001 | Benign | -1.18 | Pathogenic | 0.17 | Tolerated | 0.1586 | 0.4220 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1429A>T | M477L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M477L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The high‑accuracy methods—AlphaMissense‑Optimized, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta)—all uniformly indicate a benign effect. No prediction or folding‑stability result is missing or inconclusive. Based on the consensus of the available evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.268042 | Structured | 0.408680 | Uncertain | 0.761 | 0.250 | 0.000 | -4.772 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.19 | Likely Benign | 0.15 | Likely Benign | 0.58 | Ambiguous | 0.236 | Likely Benign | -1.25 | Neutral | 0.000 | Benign | 0.001 | Benign | -1.18 | Pathogenic | 0.17 | Tolerated | 0.1586 | 0.4220 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.2033G>A | S678N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678N is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID 6‑33441292‑G‑A). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Overall, the variant is most likely benign, and this conclusion does not contradict ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | 6-33441292-G-A | 1 | 6.20e-7 | -3.355 | Likely Benign | 0.139 | Likely Benign | Likely Benign | -0.10 | Likely Benign | 0.1 | -0.47 | Likely Benign | -0.29 | Likely Benign | 0.38 | Likely Benign | 0.067 | Likely Benign | -0.63 | Neutral | 0.001 | Benign | 0.002 | Benign | 3.43 | Benign | 0.14 | Tolerated | 3.43 | 19 | 0.1435 | 0.4863 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 2/11 « Previous | Next »